


Wie man diesen Artikel liest: Entschuldigung, dass der Text so lang und chaotisch ausgefallen ist. Um Ihre Zeit zu sparen, beginne ich jedes Kapitel mit der Einleitung "Was ich gelernt habe", in der ich in ein oder zwei Sätzen die Essenz des Kapitels zusammenfasse.
"Zeig einfach die Lösung!" Wenn Sie nur sehen möchten, zu welchem Ergebnis ich gekommen bin, wechseln Sie zum Kapitel "Ich werde einfallsreicher", aber ich denke, dass es interessanter und nützlicher ist, über die Misserfolge zu lesen.
Kürzlich wurde mir die Aufgabe übertragen, den Prozess zur Verarbeitung eines großen Volumens an DNA-Eingangssequenzen (technisch betrachtet ein SNP-Chip) einzurichten. Es war erforderlich, schnell Daten über einen bestimmten genetischen Standort (genannt SNP) zu erhalten, um sie für die anschließende Modellierung und andere Aufgaben zu nutzen. Mit Hilfe von R und AWK gelang es mir, die Daten auf natürliche Weise zu bereinigen und zu organisieren, wodurch die Verarbeitung der Anfragen erheblich beschleunigt wurde. Das war nicht einfach und erforderte zahlreiche Iterationen. Dieser Artikel wird Ihnen helfen, einige meiner Fehler zu vermeiden, und zeigt, was am Ende dabei herausgekommen ist.
Zunächst einige einführende Erklärungen.
Daten
Unser universitärer Genom-Informationszentrum hat uns die Daten in Form von 25 TB TSV zur Verfügung gestellt. Ich erhielt sie in 5 Paketen, komprimiert mit Gzip, wobei jedes Paket etwa 240 vier Gigabyte große Dateien enthielt. Jede Zeile enthielt Daten für einen SNP einer Person. Insgesamt wurden Daten für ~2,5 Millionen SNPs und ~60.000 Personen übermittelt. Neben den SNP-Informationen enthielten die Dateien zahlreiche Spalten mit Zahlen, die verschiedene Merkmale wie die Lesintensität, Häufigkeit verschiedener Allele usw. widerspiegelten. Insgesamt gab es etwa 30 Spalten mit einzigartigen Werten.
Ziel
Wie in jedem Datenmanagementprojekt war es am wichtigsten, zu bestimmen, wie die Daten genutzt werden sollen. In diesem Fall werden wir größtenteils Modelle und Arbeitsabläufe für SNP basierend auf SNP auswählen. Das heißt, wir benötigen gleichzeitig Daten nur zu einem SNP. Ich musste lernen, so einfach, schnell und kostengünstig wie möglich alle Aufzeichnungen zu extrahieren, die sich auf einen der 2,5 Millionen SNPs beziehen.
So sollte man es nicht machen
Ich zitiere ein passendes Klischee:
Ich habe nicht tausend Mal versagt, ich habe lediglich tausend Möglichkeiten gefunden, eine große Menge an Daten nicht in einem abfragbaren Format zu extrahieren.
Erster Versuch
Was ich gelernt habe: Es gibt keinen kostengünstigen Weg, 25 TB auf einmal zu extrahieren.
Nachdem ich an der Vanderbilt University den Kurs "Erweiterte Methoden der Big Data Verarbeitung" besucht hatte, war ich mir sicher, dass es einfach werden würde. Vielleicht würde es ein oder zwei Stunden dauern, um den Hive-Server einzurichten, um durch alle Daten zu laufen und die Ergebnisse zu berichten. Da unsere Daten in AWS S3 gespeichert sind, habe ich den Service , der es ermöglicht, Hive SQL-Abfragen auf S3-Daten anzuwenden, genutzt. Man muss kein Hive-Cluster einrichten und zahlt nur für die Daten, die man abruft.
Nachdem ich Athena meine Daten und deren Format gezeigt hatte, habe ich einige Tests mit ähnlichen Abfragen durchgeführt:
select * from intensityData limit 10;Und erhielt schnell gut strukturierte Ergebnisse. Fertig.
Bis wir versucht haben, die Daten in der Praxis zu verwenden…
Ich wurde gebeten, alle Informationen zu SNPs zu extrahieren, um mein Modell damit zu testen. Ich habe die Abfrage gestartet:
select * from intensityData
where snp = 'rs123456';…und wartete. Nach acht Minuten und mehr als 4 TB angeforderter Daten erhielt ich ein Ergebnis. Athena berechnet Gebühren basierend auf dem Volumen der gefundenen Daten, mit $5 pro Terabyte. Daher kostete diese einzige Anfrage $20 und acht Minuten Wartezeit. Um das Modell mit allen Daten zu durchlaufen, müsste man 38 Jahre warten und $50 Millionen zahlen. Offensichtlich war das keine Option für uns.
Parquet musste verwendet werden…
Was ich gelernt habe: Achten Sie auf die Größe Ihrer Parquet-Dateien und deren Organisation.
Zunächst versuchte ich, die Situation zu verbessern, indem ich alle TSV in konvertierte. Diese sind besonders nützlich für die Verarbeitung großer Datensätze, da die Informationen in ihnen spaltenorientiert gespeichert werden: Jede Spalte liegt in ihrem eigenen Segment im Speicher/auf der Festplatte, im Gegensatz zu Textdateien, in denen die Zeilen die Elemente jeder Spalte enthalten. Wenn man etwas finden möchte, reicht es aus, die benötigte Spalte zu lesen. Darüber hinaus wird in jeder Datei der Spalte ein Wertebereich gespeichert, sodass, wenn der gesuchte Wert nicht im Bereich der Spalte liegt, Spark keine Zeit mit dem Scannen der gesamten Datei vergeudet.
Ich startete eine einfache Aufgabe Um unsere TSV in Parquet zu konvertieren, habe ich neue Dateien in Athena hochgeladen. Das hat ungefähr 5 Stunden gedauert. Doch als ich die Abfrage startete, benötigte sie etwa genauso viel Zeit und kostete nur wenig mehr. Der Grund ist, dass Spark versuchte, die Aufgabe zu optimieren, indem es einfach einen TSV-Chunk entpackte und in seinen eigenen Parquet-Chunk speicherte. Da jeder Chunk groß genug war und die vollständigen Datensätze vieler Personen beinhaltete, waren in jeder Datei alle SNPs gespeichert. Daher musste Spark alle Dateien öffnen, um die erforderlichen Informationen zu extrahieren.
Interessanterweise ist der standardmäßig (und empfohlen) verwendete Kompressionstyp in Parquet – Snappy – nicht teilbar (splitable). Deshalb hing jeder Executor an der Aufgabe des Entpackens und des Hochladens des vollständigen Datensatzes von 3,5 GB.
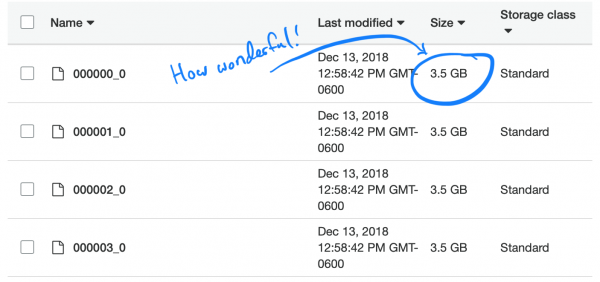
Wir analysieren das Problem
Was ich gelernt habe: sortieren ist schwierig, insbesondere wenn die Daten verteilt sind.
Ich dachte, ich hätte das Kernproblem jetzt verstanden. Ich musste die Daten nur nach der Spalte SNP sortieren und nicht nach den Personen. Dann würden in einem separaten Datenchunk mehrere SNPs gespeichert, und dann würde die "intelligente" Parquet-Funktion "nur öffnen, wenn der Wert im Bereich liegt" richtig zur Geltung kommen. Leider war es eine komplexe Aufgabe, Milliarden von Zeilen, die über den Cluster verteilt sind, zu sortieren.
Ich in der Algorithmenvorlesung an der Uni: „Ugh, es interessiert doch keinen, wie komplex die Berechnung all dieser Sortieralgorithmen ist.“
Ich versuche, in einer 20TB Tabelle nach einer Spalte zu sortieren: „Warum dauert das so lange?“ Kämpfe.
— Nick Strayer (@NicholasStrayer)
AWS möchte definitiv aus dem Grund „Ich bin ein zerstreuter Student“ kein Geld zurückgeben. Nachdem ich die Sortierung in Amazon Glue gestartet hatte, lief sie zwei Tage lang und brach dann ab.
Wie sieht es mit der Partitionierung aus?
Was ich gelernt habe: Die Partitionen in Spark sollten ausgewogen sein.
Dann kam mir die Idee, die Daten in Chromosomen zu partitionieren. Es gibt 23 davon (und ein paar mehr, wenn man die mitochondriale DNA und die nicht kartierten Bereiche berücksichtigt).
Dies würde es ermöglichen, die Daten in kleinere Portionen zu unterteilen. Wenn ich einfach eine Zeile in die Spark-Funktion zum Exportieren im Glue-Skript hinzufügen würde, partition_by = "chr", die Daten müssen in Buckets aufgeteilt werden.
Ein Gen besteht aus zahlreichen Fragmente, die Chromosomen genannt werden.
Leider hat das nicht funktioniert. Chromosomen haben unterschiedliche Größen, was bedeutet, dass sie auch unterschiedliche Mengen an Informationen enthalten. Das führt dazu, dass die Aufgaben, die Spark an die Worker gesendet hat, nicht ausgewogen waren und langsam ausgeführt wurden, weil einige Knoten früher fertig waren und untätig blieben. Aber die Aufgaben wurden erfolgreich ausgeführt. Doch bei der Abfrage eines SNP wurde die Unausgewogenheit erneut zum Problem. Die Kosten für die Verarbeitung von SNPs in größeren Chromosomen (also dort, wo wir die Daten abrufen möchten) haben sich nur etwa verzehnfacht. Viel, aber nicht genug.
Was ist, wenn wir sie in noch kleinere Partitionen unterteilen?
Was ich gelernt habe: Versuchen Sie nie, 2,5 Millionen Partitionen zu erstellen.
Ich habe beschlossen, alles auf die Spitze zu treiben und jede SNP zu partitionieren. Das garantierte eine einheitliche Größe der Partitionen. ES WAR EINE SCHLECHTE IDEE. Ich habe Glue verwendet und eine harmlose Zeile hinzugefügt partition_by = 'snp'. Die Aufgabe wurde gestartet und begann, ihre Arbeit zu verrichten. Einen Tag später überprüfte ich und sah, dass im S3 nach wie vor nichts geschrieben wurde, also stoppte ich die Aufgabe. Es scheint, dass Glue temporäre Dateien an einem versteckten Ort in S3 geschrieben hat, und es gibt viele Dateien, möglicherweise ein paar Millionen. Das Resultat meines Fehlers belief sich auf über tausend Dollar, was meinen Mentor nicht erfreute.
Partitionierung + Sortierung
Was ich gelernt habe: Das Sortieren ist nach wie vor schwierig, ebenso wie das Einrichten von Spark.
Mein letzter Versuch der Partitionierung bestand darin, die Chromosomen zu partitionieren und dann jede Partition zu sortieren. Theoretisch sollte dies jede Anfrage beschleunigen, da die gewünschten SNP-Daten innerhalb weniger Parquet-Chunks in einem bestimmten Bereich liegen sollten. Leider stellte sich das Sortieren selbst partitionierter Daten als herausfordernde Aufgabe heraus. Daher wechselte ich zu EMR für einen benutzerdefinierten Cluster und verwendete acht leistungsstarke Instanzen (C5.4xl) sowie Sparklyr, um einen flexibleren Arbeitsablauf zu schaffen…
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)…dennoch wurde die Aufgabe letztendlich nicht abgeschlossen. Ich habe es auf verschiedene Weise versucht: Ich habe den zugewiesenen Speicher für jeden Abfrageausführer erhöht, leistungsstärkere Knoten verwendet und Broadcast-Variablen eingesetzt, aber jedes Mal waren es nur halbe Lösungen, und allmählich begannen die Ausführer zu versagen, bis alles zum Stillstand kam.
Update: Es beginnt.
— Nick Strayer (@NicholasStrayer)
Ich werde einfallsreicher.
Was ich gelernt habe: Manchmal erfordern besondere Daten besondere Lösungen.
Jeder SNP hat einen Positionswert. Dies ist eine Zahl, die der Anzahl der Basen entlang seines Chromosoms entspricht. Das ist eine gute und natürliche Möglichkeit, unsere Daten zu organisieren. Zunächst wollte ich nach Bereichen jedes Chromosoms partitionieren. Zum Beispiel, Positionen 1 – 2000, 2001 – 4000 usw. Aber das Problem ist, dass SNPs ungleichmäßig über die Chromosomen verteilt sind, weshalb die Gruppengröße stark variieren wird.
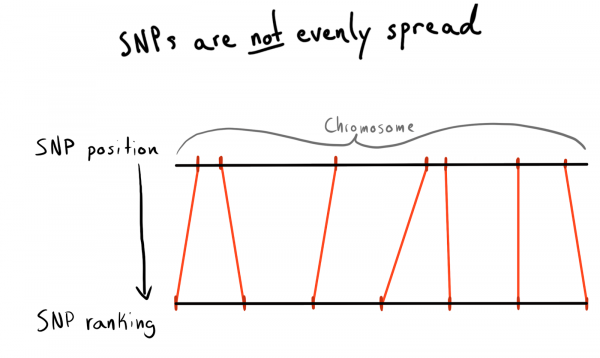
Als Ergebnis habe ich die Positionen nach Kategorien (Rang) aufgeteilt. Mit den bereits geladenen Daten habe ich eine Abfrage zur Erfassung einer Liste einzigartiger SNPs, deren Positionen und Chromosomen durchgeführt. Anschließend habe ich die Daten innerhalb jedes Chromosoms sortiert und die SNPs in Gruppen (Bins) einer bestimmten Größe gesammelt, zum Beispiel 1000 SNPs. Dies gab mir den Zusammenhang zwischen den SNPs und der Gruppe im Chromosom.
Letztendlich habe ich Gruppen (Bins) von 75 SNPs gebildet, die Gründe dafür erkläre ich weiter unten.
snp_to_bin %
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()Mein erster Versuch mit Spark
Was ich gelernt habe: Die Zusammenführung in Spark funktioniert schnell, aber das Partitionieren bleibt kostspielig.
Ich wollte dieses kleine (2,5 Millionen Zeilen) Datenframe in Spark einlesen, es mit den Rohdaten zusammenführen und dann nach der neu hinzugefügten Spalte partitionieren. bin.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
) Ich habe verwendet sdf_broadcast(), sodass Spark weiß, dass es das Datenframe an alle Knoten senden soll. Dies ist nützlich, wenn die Daten klein sind und für alle Aufgaben benötigt werden. Andernfalls versucht Spark, clever zu sein und verteilt die Daten nach Bedarf, was zu Verzögerungen führen kann.
Und wieder hat meine Idee nicht funktioniert: Die Aufgaben haben eine Zeit lang gearbeitet, das Zusammenfügen abgeschlossen, und dann begannen die ausgeführten Partitionierer, zu fehlerhaft zu werden.
Ich füge AWK hinzu
Was ich gelernt habe: Schlafen Sie nicht, während Ihnen die Grundlagen beigebracht werden. Jemand hat sicherlich Ihr Problem schon in den 1980er Jahren gelöst.
Bis zu diesem Zeitpunkt war die Ursache all meiner Misserfolge mit Spark eine Vermischung der Daten im Cluster. Möglicherweise lässt sich die Situation durch Vorverarbeitung verbessern. Ich entschied mich, die Rohtextdaten in Chromosomenspalten zu unterteilen, in der Hoffnung, Spark „vorpartitionierte“ Daten zu liefern.
Ich habe auf StackOverflow nachgeschaut, wie man nach Werten in Spalten aufteilt, und fand Mit AWK können Sie eine Textdatei nach Werten in Spalten aufteilen, indem Sie ein Skript aufzeichnen, anstatt die Ergebnisse zu senden an stdout.
Zum Ausprobieren schrieb ich ein Bash-Skript. Ich lud eine der gepackten TSV-Dateien herunter und entpackte sie mit gzip und schickte sie an awk.
gzip -dc path/to/chunk/file.gz |
awk -F 't'
'{print $1",..."$30" > "chunked/"$chr"_chr"$15".csv"}'Das hat funktioniert!
Kernfüllung
Was ich gelernt habe: gnu parallel ist eine magische Sache, die jeder nutzen sollte.
Die Aufteilung verlief ziemlich langsam, und als ich htopstartete, um die Nutzung des leistungsstarken (und teuren) EC2-Instances zu überprüfen, stellte ich fest, dass ich nur einen Kern und etwa 200 MB Speicher verwendete. Um das Problem zu lösen und nicht viel Geld zu verlieren, musste ich herausfinden, wie ich die Arbeit parallelisieren kann. Glücklicherweise fand ich in dem völlig fantastischen Buch von Jeroen Janssens ein Kapitel, das der Parallelisierung gewidmet ist. Daraus erfuhr ich von gnu parallel, einer sehr flexiblen Methode zur Implementierung von Multithreading in Unix.

Als ich die Aufteilung mit dem neuen Prozess startete, lief alles hervorragend, aber es gab einen Engpass: Der Download von S3-Objekten auf die Festplatte war nicht sehr schnell und nicht vollständig parallelisiert. Um das zu beheben, tat ich folgendes:
- Ich stellte fest, dass ich den S3-Download-Schritt direkt im Pipeline implementieren kann, wodurch die Zwischenablage auf der Festplatte vollständig ausgeschlossen wird. Das bedeutet, dass ich das Schreiben der Rohdaten auf die Festplatte vermeiden und ein noch kleineres und damit kostengünstigeres Speicherangebot bei AWS nutzen kann.
- Mit dem Befehl
aws configure set default.s3.max_concurrent_requests 50Die Anzahl der Threads, die das AWS CLI verwendet, wurde deutlich erhöht (Standard sind 10). - Ich habe auf eine Netzwerk-optimierte EC2-Instance mit dem Buchstaben n im Namen gewechselt. Ich habe festgestellt, dass der Verlust an Rechenleistung bei der Verwendung von n-Instances durch die erhöhte Ladegeschwindigkeit mehr als ausgeglichen wird. Für die meisten Aufgaben habe ich c5n.4xl verwendet.
- Ich habe gewechselt
gzipfindet man , das ist ein gzip-Tool, das coole Dinge für die Parallelisierung von ursprünglich nicht parallelisierbaren Aufgaben beim Entpacken von Dateien machen kann (das hat am wenigsten geholfen).
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe
"awk -F 't' '{print $1",..."$30">"chunked/{#}_chr"$15".csv"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneDiese Schritte wurden miteinander kombiniert, um alles sehr schnell zu machen. Dank der erhöhten Downloadgeschwindigkeit und der Vermeidung von Festplattenschreibvorgängen konnte ich jetzt ein 5-Terabyte-Paket in nur wenigen Stunden verarbeiten.
Es gibt nichts Schöneres, als alle Kerne, für die man bei AWS zahlt, auch genutzt zu sehen. Dank gnu-parallel kann ich eine 19-Gig CSV genauso schnell entpacken und aufteilen, wie ich sie herunterladen kann. Ich konnte nicht einmal Spark dazu bringen, es auszuführen.
— Nick Strayer (@NicholasStrayer)
In diesem Tweet hätte 'TSV' erwähnt werden sollen. Leider.
Verwendung der neu geparsten Daten
Was ich gelernt habe: Spark liebt unkomprimierte Daten und mag es nicht, Partitionen zu kombinieren.
Die Daten lagen nun unkomprimiert (sprich, geteilt) und teilweise sortiert in S3, und ich konnte zurück zu Spark. Es erwartete mich eine Überraschung: Ich konnte wieder nicht das gewünschte Ergebnis erzielen! Es war sehr schwierig, Spark genau zu sagen, wie die Daten partitioniert sind. Und selbst als ich das tat, stellte sich heraus, dass es zu viele Partitionen gab (95.000), und als ich mit Hilfe von coalesce die Anzahl auf ein vernünftiges Maß reduzierte, zerstörte dies meine Partitionierung. Ich bin sicher, dass das behoben werden kann, aber nach ein paar Tagen intensiver Suche fand ich keine Lösung. Letztendlich habe ich alle Aufgaben in Spark abgeschlossen, obwohl es eine Weile dauerte und meine aufgeteilten Parquet-Dateien nicht sehr klein waren (~200 Kb). Die Daten lagen jedoch genau dort, wo sie gebraucht wurden.
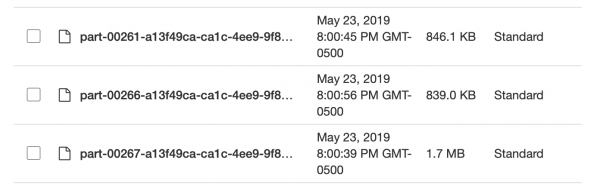
Zu klein und ungleichmäßig, wunderbar!
Testen von lokalen Spark-Abfragen
Was ich gelernt habe: In Spark gibt es zu viel Overhead beim Lösen einfacher Aufgaben.
Durch das Hochladen der Daten im durchdachten Format konnte ich die Geschwindigkeit testen. Ich habe ein Skript in R eingerichtet, um einen lokalen Spark-Server zu starten, und anschließend ein Spark-Datenrahmen aus dem angegebenen Parquet-Gruppen-Speicher geladen. Ich wollte alle Daten laden, konnte jedoch Sparklyr nicht dazu bringen, die Partitionierung zu erkennen.
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Timer starten
start_time <- Sys.time()
# Das gewünschte Bin in Spark laden
intensity_data %
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Bin nach SNP filtern und dann lokal speichern
test_subset %
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)Die Ausführung dauerte 29,415 Sekunden. Um einiges besser, aber für massenhaftes Testen noch nicht gut genug. Außerdem konnte ich die Leistung nicht durch Caching beschleunigen, da Spark immer abstürzte, wenn ich versuchte, den Datenrahmen im Speicher zu cachen, selbst als ich mehr als 50 GB Speicher für einen Datensatz zugewiesen hatte, der weniger als 15 GB groß war.
Zurück zu AWK
Was ich gelernt habe: Assoziative Arrays in AWK sind sehr effizient.
Ich hatte das Gefühl, dass ich eine höhere Geschwindigkeit erzielen konnte. Mir fiel ein, dass in dem großartigen Ich habe von einer interessanten Funktion gelesen, die "" heißt. Im Grunde genommen handelt es sich dabei um Schlüssel-Wert-Paare, die aus irgendeinem Grund in AWK anders genannt wurden, weshalb ich sie nicht besonders im Gedächtnis hatte. erinnerte mich daran, dass der Begriff „assoziative Arrays“ viel älter ist als der Begriff „Schlüssel-Wert-Paar“. Selbst wenn Sie , werden Sie diesen Begriff dort nicht finden, aber Sie werden assoziative Arrays finden! Außerdem wird „Schlüssel-Wert-Paar“ häufig mit Datenbanken assoziiert, deshalb ist es viel logischer, mit Hashmaps zu vergleichen. Ich erkannte, dass ich diese assoziativen Arrays verwenden kann, um meine SNP mit der Gruppentabelle (bin table) und Rohdaten ohne Spark zu verbinden.
Dafür verwendete ich im AWK-Skript den Block BEGIN. Das ist der Codeabschnitt, der ausgeführt wird, bevor die erste Datenzeile an den Hauptteil des Skripts übergeben wird.
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
} Der Befehl while(getline...) lädt alle Zeilen aus der CSV-Gruppe (bin), wobei die erste Spalte (Name des SNP) als Schlüssel für das assoziative Array festgelegt wird. bin und der zweite Wert (Gruppe) als Wert. Dann im Block { }, der auf alle Zeilen der Hauptdatei angewendet wird, wird jede Zeile in eine Ausgabedatei gesendet, die einen eindeutigen Namen je nach ihrer Gruppe (bin) erhält: ..._bin_"bin[$1]"_....
Variablen batch_num und chunk_id entsprachen den vom Pipelining bereitgestellten Daten, wodurch ein Zustand von Rennen vermieden wurde und jeder Ausführungssthread, der gestartet wurde parallel, schrieb in seine eigene einzigartige Datei.
Da ich alle Rohdaten nach Chromosomen aufgeteilt habe, die aus meinem vorherigen Experiment mit AWK übrig geblieben sind, konnte ich jetzt ein weiteres Bash-Skript schreiben, um chromosomenweise zu verarbeiten und tiefere partitionierte Daten nach S3 zu übergeben.
DESIRED_CHR='13'
# Chromosomendaten von s3 herunterladen und in Bins aufteilen
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'lesen von {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=""$DESIRED_CHR"" -v chunk="{}" -f split_on_chr_bin.awk"
# Kombiniere alle parallel verarbeiteten Chunks zu einer Datei und lade sie mit R in rds hoch
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/* Im Skript gibt es zwei Abschnitte parallel.
Im ersten Abschnitt werden die Daten aus allen Dateien gelesen, die Informationen über das benötigte Chromosom enthalten. Anschließend werden diese Daten auf mehrere Threads verteilt, die die Dateien in die entsprechenden Gruppen (Binsen) sortieren. Um Rennbedingungen zu vermeiden, bei denen mehrere Threads in eine Datei schreiben, überträgt AWK die Dateinamen zur Speicherung der Daten an unterschiedliche Orte, zum Beispiel chr_10_bin_52_batch_2_aa.csv. Infolgedessen entstehen auf der Festplatte viele kleine Dateien (dafür habe ich terabyte-große EBS-Volumen verwendet).
Die Pipeline im zweiten Abschnitt parallel geht durch die Gruppen (Binsen) und vereint ihre einzelnen Dateien in gemeinsame CSV mit cat, und exportiert sie anschließend.
Transcoding nach R?
Was ich gelernt habe: kann von stdin und stdout aus einem R-Skript aufgerufen werden, wodurch es auch im Pipeline-Prozess verwendet werden kann.
Im Bash-Skript haben Sie möglicherweise eine solche Zeile bemerkt: ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... Sie überträgt alle verketteten Dateien der Gruppe (Binsen) in das nachfolgende R-Skript. {} ist eine spezielle Methode parallel, die alle an den angegebenen Stream gesendeten Daten direkt in den Befehl einfügt. Die Option {#} stellt eine eindeutige ID für den Ausführungsstrom bereit, während {%} stellt die Auftragsplatznummer dar (wird wiederholt, aber nie gleichzeitig). Eine Liste aller Optionen finden Sie in
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
) Wenn die Variable file("stdin") an readr::read_csv, werden die in das R-Skript übertragenen Daten in einen Data Frame geladen, der dann als .rds-Datei mit Hilfe von aws.s3 direkt in S3 geschrieben wird.
RDS ist eine Art vereinfachte Version von Parquet, ohne die Raffinessen eines Spalten-Speichers.
Nach Abschluss des Bash-Skripts erhielt ich eine Reihe von .rds-Dateien, die in S3 liegen, was mir ermöglichte, effizientes Komprimieren und eingebaute Typen zu nutzen.
Trotz der Verwendung von langsamem R funktionierte alles sehr schnell. Es ist nicht überraschend, dass die Teile in R, die für das Lesen und Schreiben von Daten zuständig sind, gut optimiert sind. Nach Tests mit einem mittelgroßen Chromosom wurde der Auftrag auf einer C5n.4xl Instanz in etwa zwei Stunden abgeschlossen.
S3-Beschränkungen: Dank der intelligenten Implementierung von Pfaden kann S3 viele Dateien verarbeiten.
Was ich gelernt habeIch machte mir Sorgen, ob S3 die vielen ihr übergebenen Dateien verarbeiten kann. Ich konnte die Dateinamen sinnvoll gestalten, aber wie wird S3 danach suchen?
Ich machte mir Sorgen, ob S3 mit der großen Anzahl an übergebenen Dateien umgehen kann. Ich könnte die Dateinamen sinnvoll gestalten, aber wie wird S3 danach suchen?

Ordner in S3 dienen lediglich der Ästhetik; tatsächlich interessiert das System das Zeichen nicht. /.
Es scheint, dass S3 den Pfad zu einer bestimmten Datei als einfachen Schlüssel in einer Art Hash-Tabelle oder einer dokumentenbasierten Datenbank darstellt. Ein Bucket kann als Tabelle angesehen werden, während Dateien als Einträge in dieser Tabelle fungieren.
Da Geschwindigkeit und Effizienz entscheidend für den Gewinn bei Amazon sind, ist es nicht überraschend, dass dieses "Schlüssel-als-Pfad-zur-Datei"-System hervorragend optimiert ist. Ich habe versucht, ein Gleichgewicht zu finden: Es sollten nicht viele GET-Anfragen nötig sein, aber die Anfragen sollten schnell ausgeführt werden. Es stellte sich heraus, dass es am besten ist, etwa 20.000 Bin-Dateien zu erstellen. Ich denke, wenn man die Optimierung weiterführt, kann man die Geschwindigkeit steigern (zum Beispiel durch die Erstellung eines speziellen Buckets nur für Daten, wodurch die Größe der Suchtabelle reduziert wird). Aber für weitere Experimente fehlte bereits die Zeit und das Geld.
Wie sieht es mit der Kompatibilität aus?
Was ich gelernt habe: Der Hauptgrund für Zeitverlust ist die vorzeitige Optimierung Ihrer Speicherungsmethode.
In diesem Moment ist es sehr wichtig, sich zu fragen: „Warum ein proprietäres Dateiformat verwenden?“ Der Grund liegt in der Ladegeschwindigkeit (komprimierte gzip CSV-Dateien benötigten siebenmal länger) und der Kompatibilität mit unseren Arbeitsabläufen. Ich könnte meine Entscheidung überdenken, wenn R in der Lage ist, Parquet- (oder Arrow-) Dateien problemlos ohne die Belastung durch Spark zu laden. In unserem Labor arbeitet jeder mit R, und falls ich Daten in ein anderes Format umwandeln muss, habe ich immer noch die ursprünglichen Textdaten, sodass ich einfach die Pipeline erneut ausführen kann.
Arbeitsteilung
Was ich gelernt habe: Versuchen Sie nicht, Aufgaben manuell zu optimieren; überlassen Sie das dem Computer.
Ich habe den Workflow für ein Chromosom optimiert, jetzt müssen die restlichen Daten bearbeitet werden.
Ich wollte mehrere EC2-Instanzen für die Umwandlung starten, hatte jedoch Bedenken, eine extrem unausgeglichene Last in den verschiedenen Verarbeitungsaufgaben zu erhalten (ähnlich wie Spark unter unausgeglichenen Partitionen litt). Außerdem wollte ich nicht für jedes Chromosom eine einzelne Instanz hochfahren, da es in AWS-Accounts eine Standardbeschränkung von 10 Instanzen gibt.
Daraufhin beschloss ich, ein R-Skript zur Optimierung der Verarbeitungsaufgaben zu schreiben.
Zuerst bat ich S3, auszurechnen, wie viel Speicherplatz jede Chromosom in Anspruch nimmt.
library(aws.s3)
library(tidyverse)
chr_sizes %
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Chromosom aus dem Dateinamen extrahieren
chr = str_extract(Key, 'chr.{1,4}.csv') %>%
str_remove_all('chr|.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Dividiere, um den Wert in GB zu erhalten
# Ein tibble: 27 x 2
chr total_size
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … mit 17 weiteren Zeilen Anschließend schrieb ich eine Funktion, die die Gesamtsumme nimmt, die Reihenfolge der Chromosomen mischt und sie in Gruppen einteilt. num_jobs und angibt, wie stark die Größen aller Verarbeitungsaufgaben variieren.
num_jobs <- 7
# Wie groß wäre jede Aufgabe, wenn sie perfekt aufgeteilt wäre?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job %
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)
# Ein tibble: 1 x 2
sd data
1 153.Dann habe ich mit purrr tausend Mischungen durchgeführt und das beste ausgewählt.
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1) So erhielt ich eine Reihe von Aufgaben, die in ihrer Größe sehr ähnlich waren. Dann blieb nur noch, mein vorheriges Bash-Skript in eine große Schleife zu packen. für. Für die Erstellung dieser Optimierung benötigte ich etwa 10 Minuten. Das ist bei weitem weniger, als ich für die manuelle Erstellung von Aufgaben aufgewendet hätte, falls sie unausgewogen gewesen wären. Daher denke ich, dass ich mit dieser vorläufigen Optimierung nicht falsch lag.
for DESIRED_CHR in "16" "9" "7" "21" "MT"
do
# Code zur Verarbeitung eines einzelnen Chromosoms
fiAm Ende füge ich den Abschaltbefehl hinzu:
sudo shutdown -h now … und alles hat geklappt! Mit AWS CLI habe ich Instanzen hochgefahren und über die Option user_data ihnen Bash-Skripte für ihre Bearbeitungsaufgaben übergeben. Diese wurden ausgeführt und automatisch abgeschaltet, sodass ich nicht für überschüssige Rechenleistung bezahlen musste.
aws ec2 run-instances ...
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]"
--user-data file://<<job_script_loc>>Packen wir es ein!
Was ich gelernt habe: API sollte einfach sein, um Einfachheit und Flexibilität in der Nutzung zu gewährleisten.
Endlich habe ich die Daten in der gewünschten Form und am richtigen Ort erhalten. Jetzt galt es, den Prozess der Datennutzung so einfach wie möglich zu gestalten, damit es für meine Kollegen leichter wird. Ich wollte eine einfache API zur Erstellung von Anfragen entwickeln. .rds Wenn ich mich in Zukunft für Parquet-Dateien entscheiden sollte, sollte dies kein Problem für mich und meine Kollegen darstellen. Daher entschied ich mich, ein internes R-Paket zu erstellen.
Ich habe ein sehr einfaches Paket erstellt und dokumentiert, das nur einige Funktionen zur Datenabfrage umfasst, die sich um die Funktion get_snpgruppieren. Zudem habe ich für meine Kollegen eine Webseite eingerichtet, damit sie problemlos Beispiele und Dokumentation einsehen können.
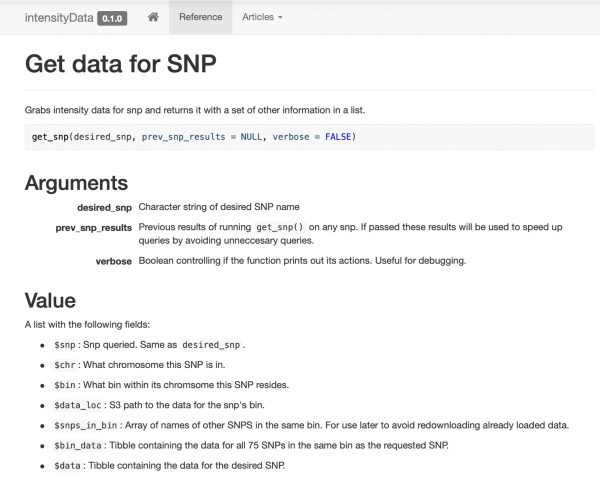
Intelligentes Caching
Was ich gelernt habe: Wenn Ihre Daten gut aufbereitet sind, lässt sich das Caching problemlos umsetzen!
Da einer der Hauptarbeitsabläufe dasselbe Analysemodell auf das SNP-Paket anwendete, beschloss ich, die Gruppierung (Binning) zu meinem Vorteil zu nutzen. Bei der Übertragung der Daten über SNP wird auch alle Informationen aus der Gruppe (Bin) an das zurückgegebene Objekt angehängt. Theoretisch könnten also alte Anfragen die Verarbeitung neuer Anfragen beschleunigen.
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
... Bei der Paketierung habe ich viele Benchmarks durchgeführt, um die Geschwindigkeit verschiedener Methoden zu vergleichen. Ich empfehle, dies nicht zu vernachlässigen, da die Ergebnisse manchmal überraschend sein können. Zum Beispiel, dplyr::filter hat sich als deutlich schneller erwiesen als die Zeilenaufnahme mittels Indexierung, und das Abrufen einer Spalte aus dem gefilterten Data Frame funktionierte erheblich schneller als die Anwendung der Indexierungssyntax.
Beachten Sie, dass das Objekt prev_snp_results den Schlüssel snps_in_bin. Dies ist ein Array aller einzigartigen SNPs in der Gruppe (bin), das eine schnelle Überprüfung ermöglicht, ob bereits Daten aus der vorherigen Abfrage vorhanden sind. Es vereinfacht zudem die iterativen Durchläufe über alle SNPs in der Gruppe (bin) mit diesem Code:
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}Ergebnisse
Jetzt können wir (und haben ernsthaft begonnen), Modelle und Szenarien zu testen, die uns zuvor nicht zugänglich waren. Das Beste daran ist, dass meine Kollegen im Labor sich nicht um verschiedene Komplikationen kümmern müssen. Sie haben einfach eine funktionierende Funktion.
Und obwohl das Paket sie von den Einzelheiten befreit, habe ich versucht, das Datenformat einfach genug zu gestalten, damit sie damit umgehen können, falls ich morgen plötzlich verschwinde...
Die Geschwindigkeit hat sich deutlich verbessert. Üblicherweise scannen wir funktional bedeutende Fragmente des Genoms. Früher war das nicht möglich (es war zu teuer), aber jetzt, dank der Gruppenstruktur (Bins) und Caching, benötigt die Abfrage eines SNP im Durchschnitt weniger als 0,1 Sekunden, und der Datennutzung ist so gering, dass die Kosten für S3 äußerst niedrig sind.
Kürzlich wurde ich damit beauftragt, über 25 TB roher Genotypdaten für mein Labor zu verwalten. Als ich anfing, dauerte die Abfrage eines SNPs mit Spark 8 Minuten und kostete $20. Nach der Verwendung von AWK + schnellere Bearbeitung benötigt es jetzt weniger als eine Zehntelsekunde und kostet $0,00001. Mein persönlicher Erfolg.
— Nick Strayer (@NicholasStrayer)
Fazit
Dieser Artikel ist kein Leitfaden. Die Lösung ist individuell und wahrscheinlich nicht optimal. Vielmehr handelt es sich um eine Erzählung über meine Reise. Ich möchte, dass andere verstehen, dass solche Lösungen nicht einfach vollständig im Kopf entstehen, sondern das Ergebnis von Versuch und Irrtum sind. Außerdem, wenn Sie einen Experten für Datenanalyse suchen, bedenken Sie, dass für die effektive Nutzung dieser Werkzeuge Erfahrung erforderlich ist, und Erfahrung kostet Geld. Ich bin glücklich, dass ich die Mittel hatte, aber viele andere, die die gleiche Arbeit besser machen könnten als ich, werden nie die Gelegenheit dazu haben, weil ihnen das Geld fehlt, selbst um es zu versuchen.
Die Werkzeuge für Big Data sind universell. Wenn Sie Zeit haben, können Sie wahrscheinlich eine schnellere Lösung erstellen, indem Sie eine "clevere" Datenbereinigung, Speicherung und Extraktionsmethoden anwenden. Letztendlich kommt es darauf an, die Kosten und den Nutzen abzuwägen.
Was ich gelernt habe:
- Es gibt keinen günstigen Weg, 25 TB auf einmal zu parsen;
- Achten Sie auf die Größe Ihrer Parquet-Dateien und deren Organisation;
- Partitionen in Spark müssen ausgeglichen sein;
- Versuchen Sie niemals, 2,5 Millionen Partitionen zu erstellen;
- Es ist weiterhin schwierig, alles zu sortieren und Spark zu konfigurieren;
- Manchmal erfordern spezielle Daten spezielle Lösungen;
- Die Zusammenführung in Spark funktioniert schnell, aber die Partitionierung bleibt teuer;
- Schlaf nicht ein, wenn dir die Grundlagen beigebracht werden—jemand hat dein Problem vielleicht schon in den 1980ern gelöst;
gnu parallelEs ist eine magische Sache, die jeder nutzen sollte;- Spark mag unkomprimierte Daten und liebt es nicht, Partitionen zu kombinieren;
- In Spark gibt es zu viele Overheads bei der Lösung einfacher Aufgaben;
- Assoziative Arrays in AWK sind sehr effizient;
- Man kann auf
stdinundstdoutaus einem R-Skript zugreifen und es somit in der Pipeline verwenden; - Dank der cleveren Implementierung von S3 kann es viele Dateien verarbeiten;
- Der Hauptgrund für Zeitverlust ist die vorzeitige Optimierung deiner Speichermethode;
- Versuche nicht, Aufgaben manuell zu optimieren, lass das den Computer erledigen;
- Die API sollte einfach sein, um Benutzerfreundlichkeit und Flexibilität zu gewährleisten;
- Wenn deine Daten gut vorbereitet sind, wird das Caching einfach sein!
Quelle: habr.com
