


In der aktuellen COVID-19-Pandemie sind viele Probleme aufgetreten, auf die Hacker gerne aufmerksam wurden. Von mit 3D-Druckern hergestellten Gesichtsschutzschilden und selbstgemachten medizinischen Masken bis hin zur Ersetzung von vollständigen mechanischen Beatmungsgeräten – dieser Ideenstrom hat inspiriert und begeistert. Gleichzeitig gab es Versuche, sich in einem anderen Bereich voranzutreiben: der Forschung, die darauf abzielt, gegen das Virus selbst zu kämpfen.
Offenbar hat der Ansatz, der versucht, das Problem an der Wurzel zu packen, das größte Potenzial, um die aktuelle Pandemie zu stoppen und zukünftigen zuvorzukommen. Dieser Ansatz, der zum Bereich „Kenne deinen Feind“ gehört, wird im Rechenprojekt Folding@Home vertreten. Millionen von Menschen haben sich für das Projekt registriert und spenden einen Teil der Rechenleistung ihrer Prozessoren und GPUs, wodurch der größte [verteilte] Supercomputer in der Geschichte entstanden ist.
Aber wozu genau werden all diese Exaflops eingesetzt? Warum sollten solche Rechenleistungen in die ? Какая тут работает биохимия, зачем вообще белкам нужно укладываться? Вот краткий обзор фолдинга белков: что это, как он происходит и в чём его важность.
Zunächst das Wichtigste: Warum sind Proteine notwendig?
Proteine sind lebensnotwendige Strukturen. Sie liefern nicht nur das Baumaterial für Zellen, sondern fungieren auch als Enzyme, die praktisch alle biochemischen Reaktionen katalysieren. Proteine, ob sie oder , bestehen aus langen Ketten , die in einer bestimmten Reihenfolge angeordnet sind. Die Funktionen von Proteinen hängen davon ab, welche Aminosäuren an bestimmten Stellen des Proteins angeordnet sind. Wenn ein Protein beispielsweise mit einem positiv geladenen Molekül interagieren muss, sollte der Verbindungsort mit negativ geladenen Aminosäuren gefüllt sein.
Um zu verstehen, wie Proteine die Struktur erhalten, die ihre Funktion bestimmt, müssen wir die Grundlagen der Molekularbiologie und den Informationsfluss in der Zelle durchgehen.
Die Produktion oder von Proteinen beginnt mit dem Prozess der . Während der Transkription wickelt sich die Doppelhelix der DNA, die die genetischen Informationen der Zelle enthält, teilweise ab und ermöglicht dem Enzym namens Die Aufgabe der RNA-Polymerase besteht darin, eine RNA-Kopie oder Transkription eines Gens zu erstellen. Diese Genkopie, die als bezeichnet wird, ist ein einzelnes Molekül, das perfekt dafür geeignet ist, die intrazellulären Proteinfabriken, , die für die Produktion oder von Proteinen verantwortlich sind, zu steuern.
Ribosomen fungieren wie Montagegeräte – sie erfassen die mRNA-Vorlage und ordnen sie anderen kleinen RNA-Stücken zu, (Transfer-RNA). Jede tRNA hat zwei aktive Bereiche – einen Abschnitt von drei Basen, der als bezeichnet wird und mit den entsprechenden Codons der mRNA übereinstimmen muss, sowie einen Bereich zur Bindung der für dieses spezifischen Aminosäure. Während der Translation versuchen die tRNA-Moleküle im Ribosom zufällig, sich mit der mRNA unter Verwendung der Anticodons zu verbinden. Bei Erfolg fügt das tRNA-Molekül seine Aminosäure der vorherigen hinzu und bildet das nächste Glied in der Kette von Aminosäuren, die durch die mRNA kodiert sind.
Diese Aminosäuresequenz bildet die erste Ebene der strukturellen Hierarchie eines Proteins, weshalb sie auch als dessen . Die gesamte dreidimensionale Struktur eines Proteins und seine Funktionen hängen direkt von der primären Struktur ab und sind von den unterschiedlichen Eigenschaften jeder Aminosäure und deren Wechselwirkungen untereinander abhängig. Wären diese chemischen Eigenschaften und Wechselwirkungen der Aminosäuren nicht vorhanden, würden immer noch lineare Sequenzen ohne dreidimensionale Struktur bleiben. Dies kann man jedes Mal beim Kochen beobachten – in diesem Prozess findet eine thermische der dreidimensionalen Struktur von Proteinen statt.
Langreichweitige Bindungen der Proteinbestandteile
Der nächsten Ebene der dreidimensionalen Struktur, die über die primäre hinausgeht, wurde der raffinierte Name gegeben. Sie umfasst Wasserstoffbindungen zwischen Aminosäuren, die relativ nah beieinanderliegen. Der Kern dieser stabilisierenden Wechselwirkungen lässt sich auf zwei Aspekte reduzieren: und . Die Alpha-Helix bildet einen eng gewundenen Abschnitt des Polypeptids, während das Beta-Faltblatt ein glattes und breites Gebiet darstellt. Beide Strukturen haben sowohl strukturelle als auch funktionale Eigenschaften, die von den Eigenschaften der sie bildenden Aminosäuren abhängen. Zum Beispiel, wenn die Alpha-Helix hauptsächlich aus hydrophilen Aminosäuren besteht, wie oder , dann wird sie wahrscheinlich an wässrigen Reaktionen teilnehmen.
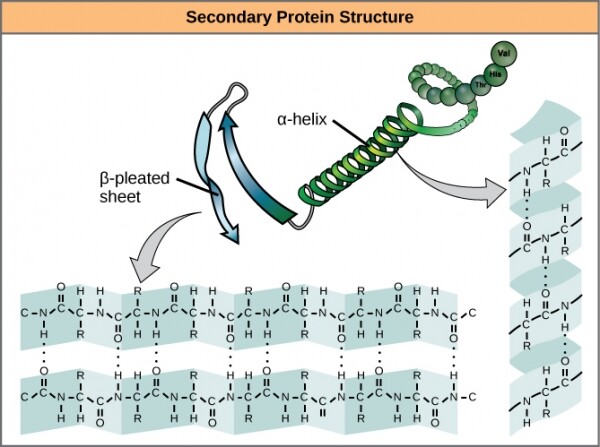
Alpha-Helices und Beta-Faltblätter in Proteinen. Wasserstoffbrückenbindungen entstehen während der Proteinexpression.
Diese beiden Strukturen und ihre Kombinationen bilden die nächste Ebene der Proteinstruktur — . Im Gegensatz zu einfachen Fragmenten der sekundären Struktur wird die tertiäre Struktur hauptsächlich von der Hydrophobizität beeinflusst. In den Zentren der meisten Proteine befinden sich Aminosäuren mit hoher Hydrophobizität, wie oder , und Wasser wird aufgrund der „fetten“ Natur der Radikale von dort ausgeschlossen. Diese Strukturen treten häufig in transmembranären Proteinen auf, die in die doppelte Lipidmembran integriert sind, die die Zellen umgibt. Die hydrophoben Abschnitte der Proteine bleiben thermodynamisch stabil innerhalb des fetthaltigen Teils der Membran, während die hydrophilen Abschnitte des Proteins der wässrigen Umgebung auf beiden Seiten ausgesetzt sind.
Auch die Stabilität der tertiären Strukturen wird durch langreichweitige Bindungen zwischen den Aminosäuren gewährleistet. Ein klassisches Beispiel für solche Bindungen ist , die häufig zwischen zwei Cysteinradikalen auftritt. Wenn Sie während einer Perma-Behandlung im Friseursalon den Geruch von etwas, das ein wenig nach faulen Eiern riecht, wahrgenommen haben, dann war dies eine partielle Denaturierung der tertiären Struktur des in den Haaren enthaltenen Keratins, die durch die Reduktion der Disulfidbindungen mithilfe von enthält, die Schwefel Gemischen.
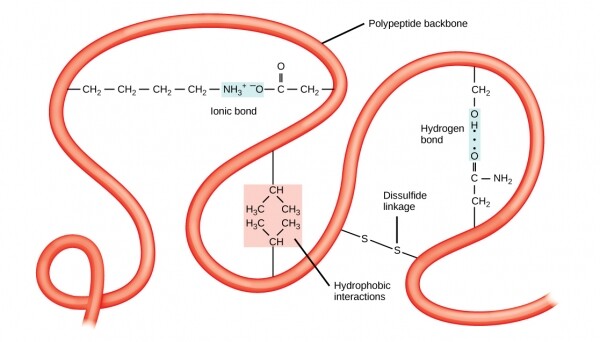
Die tertiäre Struktur wird durch langreichweitige Wechselwirkungen stabilisiert, wie Hydrophobizität oder Disulfidbindungen.
Disulfidbindungen können zwischen Radikalen in einer Polypeptidkette oder zwischen Cysteinresten aus verschiedenen vollständigen Ketten. Die Wechselwirkungen zwischen verschiedenen Ketten bilden Struktur von Proteinen. Ein hervorragendes Beispiel für eine quartäre Struktur ist in Ihrem Blut. Jedes Hämoglobinkohlenstoffmolekül besteht aus vier identischen Globinen, die Teile eines Proteins sind, wobei jeder durch Disulfidbrücken an einer bestimmten Stelle im Polypeptid gehalten wird und mit einem Häm-Molekül verbunden ist, das Eisen enthält. Alle vier Globine sind durch intermolekulare Disulfidbrücken verbunden, und das gesamte Molekül kann gleichzeitig mit mehreren Luftmolekülen, bis zu vier, interagieren und diese bei Bedarf freisetzen.
Die Modellierung von Strukturen auf der Suche nach einer Heilung für Krankheiten
Polypeptidketten beginnen während der Translation, ihre endgültige Form einzunehmen, wenn die wachsende Kette aus der Ribosomen austritt – ähnlich wie ein Drahtstück aus einer Legierung mit Formgedächtnis komplexe Formen bei Erwärmung annehmen kann. Doch wie immer in der Biologie ist nicht alles so einfach.
In vielen Zellen wird vor der Translation die transkribierte Gene einer erheblichen Bearbeitung unterzogen, die die Grundstruktur des Proteins im Vergleich zur reinen Basenfolge des Gens deutlich verändert. Dabei greifen die Translationsmechanismen häufig auf molekulare Begleiter zurück, Proteine, die vorübergehend an die entstehende Peptidkette binden und ihr nicht erlauben, eine Zwischenform anzunehmen, von der sie dann nicht mehr zur endgültigen Form übergehen können.
Das bringt uns zu der Erkenntnis, dass die Vorhersage der endgültigen Form von Proteinen keine triviale Aufgabe ist. Jahrzehntelang war die Röntgenkristallographie die einzige physikalische Methode zur Untersuchung der Proteinstruktur. Erst Ende der 1960er Jahre begannen biophysikalische Chemiker, computergestützte Modelle für das Falten von Proteinen zu entwickeln, wobei der Schwerpunkt hauptsächlich auf der Modellierung der sekundären Struktur lag. Diese Methoden und ihre Nachfolger benötigen enorme Mengen an Eingabedaten zusätzlich zur primären Struktur – zum Beispiel Tabellen mit Bindungswinkeln von Aminosäuren, Listen der Hydrophobizität, geladene Zustände und sogar die Erhaltung der Struktur und Funktionsweise über evolutionäre Zeiträume hinweg – alles, um zu erahnen, wie das endgültige Protein aussehen wird.
Die heutigen computergestützten Methoden zur Vorhersage der Sekundärstruktur, die insbesondere im Netzwerk Folding@Home eingesetzt werden, erreichen eine Genauigkeit von etwa 80 % – was angesichts der Komplexität der Aufgabe recht gut ist. Die von den Vorhersagemodellen erhaltenen Daten für Proteine wie das Spike-Protein von SARS-CoV-2 werden mit den Daten aus physikalischen Studien des Virus verglichen. Letztendlich wird es möglich sein, die genaue Struktur des Proteins zu bestimmen und möglicherweise zu verstehen, wie das Virus an die Rezeptoren bindet. des Menschen in den Atemwegen, die ins Innere des Körpers führen. Wenn wir diese Struktur entschlüsseln können, werden wir wahrscheinlich Medikamente finden, die die Bindung blockieren und eine Infektion verhindern.
Forschung zu Protein-Faltung steht im Zentrum unseres Verständnisses für eine Vielzahl von Erkrankungen und Infektionen. Selbst wenn wir mit Hilfe des Folding@Home-Netzwerks herausfinden, wie wir COVID-19 besiegen können, dessen exponentielles Wachstum wir in letzter Zeit beobachten, wird dieses Netzwerk nicht lange ohne Arbeiten bleiben. Es ist ein Forschungswerkzeug, das sich bestens für das Studium von Proteinmodellen eignet, die den Grundlagen von Dutzenden von Krankheiten zugrunde liegen, die mit fehlerhafter Protein-Faltung verbunden sind, wie etwa Alzheimer oder einer Variante der Creutzfeldt-Jakob-Krankheit, die oft fälschlicherweise als „Rinderwahn“ bezeichnet wird. Und wenn unweigerlich ein weiterer Virus auftritt, werden wir bereits bereit sein, ihn erneut zu bekämpfen.
Quelle: habr.com
