

Der Traum eines jeden Menschen ist es, so lange wie möglich jung zu bleiben. Wir wollen nicht altern und krank werden, fürchten uns vor allem – Krebs, Alzheimer, Herzinfarkt, Schlaganfall… Es ist an der Zeit, herauszufinden, woher der Krebs kommt, ob es Zusammenhänge zwischen Herzinsuffizienz und Alzheimer, Unfruchtbarkeit und Hörverlust gibt. Warum können antioxidative Ergänzungen manchmal mehr schaden als nützen? Und das Wichtigste: Können wir lange und gesund leben, und wenn ja, wie?
In unserem Körper arbeiten winzige "Energiezentralen" – die Mitochondrien. Sie sind verantwortlich für unsere Gesundheit und unser Wohlbefinden. Wenn sie gut arbeiten, mangelt es uns nicht an Energie. Wenn sie jedoch schlecht arbeiten, leiden wir unter Krankheiten. Dr. Lee Now enthüllt das Geheimnis: Erkrankungen, die auf den ersten Blick nicht miteinander verwandt erscheinen – Diabetes, Krebs, Schizophrenie, chronische Erschöpfung, Parkinson und andere – haben eine gemeinsame Ursache.
Heute wissen wir, wie wir die Funktion der Mitochondrien, die unseren Körper zu 90 % mit Energie versorgen, verbessern können. In diesem Buch finden Sie aktuelle Informationen zu Ernährung, Lebensstil, ketogener Diät und Nahrungsergänzungsmitteln, die die Gesundheit der Mitochondrien wiederherstellen und somit auch unser Wohlbefinden fördern.
Ein Auszug. Mitochondriales Syndrom
Es fällt mir schwer, das zuzugeben, aber ich war Zuschauer der Reality-Show "Der Bachelor". Besonders beeindruckte mich die dritte Episode der 17. Staffel (Januar 2013), in der Sin (der Bachelor) und Ashley (eine Kandidatin) ein Treffen mit zwei Mädchen hatten, die an einer Mitochondrienkrankheit leiden. Für viele von Ihnen, die die Episode gesehen haben, war das wahrscheinlich die erste Begegnung mit dem mitochondrialen Syndrom (das mitochondriale Syndrom ist ein Komplex von Erkrankungen, die mit genetischen Schäden an den Mitochondrien verbunden sind). Diese Krankheitsgruppe wird jedoch immer gründlicher erforscht, da genetische Testtechnologien und Genomsequenzierung einfacher, günstiger und zugänglicher werden.
Bis zu den 80er Jahren des letzten Jahrhunderts, als das menschliche Mitrochondrien-Genom vollständig sequenziert wurde, waren Berichte über mitochondriale Krankheiten selten. Diese Situation änderte sich mit der Möglichkeit, die mtDNA vieler Patienten zu entschlüsseln. Dies führte zu einem dramatischen Anstieg der registrierten Patienten, die an erblichen mitochondrialen Erkrankungen leiden. Dazu gehört ungefähr einer von fünf (oder sogar zweieinhalb) tausend Menschen. Hierbei berücksichtigen wir nicht Personen mit unauffälligen Formen mitochondrialer Krankheiten. Zudem ist auch die Liste der Symptome des mitochondrialen Syndroms stark gewachsen, was auf die chaotische Natur dieser Erkrankungen hinweist.
Mitochondriale Erkrankungen zeichnen sich durch äußerst komplexe genetische und klinische Bilder aus, die eine Mischung aus einem sehr breiten Spektrum bestehender diagnostischer Kategorien darstellen. Die Vererbungsmuster unterliegen manchmal, manchmal jedoch auch nicht den Mendelschen Gesetzen. Mendel beschrieb die Gesetzmäßigkeiten der Vererbung von Merkmalen über normale Gene der Kern-DNA. Die Wahrscheinlichkeit des Vorliegens eines genetischen Merkmals oder einer erblichen Erkrankung kann leicht anhand einer quantitativen Vorhersage der Aufspaltungseffekte der Nachkommen hinsichtlich verschiedener qualitativer Merkmale durch die zufällige Vererbung einer von zwei Kopien desselben Gens von jedem Elternteil berechnet werden (in der Folge erhält jeder Nachkomme zwei Kopien jedes Gens). In Fällen, in denen das mitochondriale Syndrom durch einen Defekt in den Kern-Genen verursacht wird, folgen die entsprechenden Vererbungsmuster tatsächlich den Mendelschen Regeln. Es gibt jedoch zwei Arten von Genomen, die das Funktionieren der Mitochondrien gewährleisten: mitochondrialer DNA (nur mütterlicherseits übertragen) und kernhaltiger DNA (von beiden Eltern geerbt). Infolgedessen variieren die Vererbungstypen von autosomal-dominant bis autosomal-rezessiv sowie durch die mütterliche Vererbung des genetischen Materials.
Die Situation wird zusätzlich dadurch kompliziert, dass zwischen mtDNA und nukleärer DNA komplexe Wechselwirkungen auftreten. Infolgedessen können dieselben mtDNA-Mutationen bei Geschwistern, die in derselben Familie leben (sie können unterschiedliche nukleäre DNA und identische mtDNA haben), stark unterschiedliche Symptome hervorrufen, während Mutationen identische Symptome verursachen können. Sogar bei Zwillingen mit derselben Diagnose können die klinischen Bilder der Krankheit radikal unterschiedlich sein (die spezifischen Symptome hängen davon ab, welche Gewebe durch den pathogenen Prozess betroffen sind), während Menschen mit Mutationen ähnliche Symptome aufweisen können, die ein ähnliches Krankheitsbild ergeben.
Unabhängig davon gibt es in der maternalen Eizelle eine Vielzahl von mtDNA-Variationen, was sämtliche Vorhersagen bezüglich der Ergebnisse der genetischen Vererbung in Frage stellt. Die Natur dieser Krankheitsgruppe ist so chaotisch, dass das Set von Symptomen, die mit diesen Erkrankungen verbunden sind, von Jahrzehnt zu Jahrzehnt variieren kann und selbst bei Geschwistern mit identischen Mutationen der mitochondriellen DNA unterschiedlich ist. Darüber hinaus kann das mitochondriale Syndrom manchmal einfach verschwinden, obwohl es vererbt wurde (oder vererbt werden sollte). Doch solche glücklichen Fälle sind selten; meist schreiten mitochondriale Erkrankungen fort. In den Tabellen 2.2 und 2.3 sind die Krankheiten und Symptome aufgeführt, die mit mitochondrialer Dysfunktion zusammenhängen, sowie die genetischen Faktoren dieser Erkrankungen. Der Wissenschaft sind derzeit über 200 Typen von Mitochondrienmutationen bekannt. Forschungsergebnisse zeigen, dass viele degenerative Krankheiten durch derartige Mutationen verursacht werden (das bedeutet, dass wir eine große Anzahl von Erkrankungen neu klassifizieren müssen, indem wir sie in die Kategorie der mitochondrialen Krankheiten überführen).
Wie bekannt ist, können diese Mutationen dazu führen, dass Mitochondrien ihre Funktion der Energieproduktion einstellen, wodurch die Zellen ihre Arbeit unterbrechen oder sterben können. Alle Zellen (außer roten Blutkörperchen) enthalten Mitochondrien, und daher betrifft das mitochondriale Syndrom verschiedene, multifunktionale Systeme des Körpers (gleichzeitig oder nacheinander).
Tabelle 2.2. Anzeichen, Symptome und Krankheiten, die durch mitochondriale Dysfunktion verursacht werden
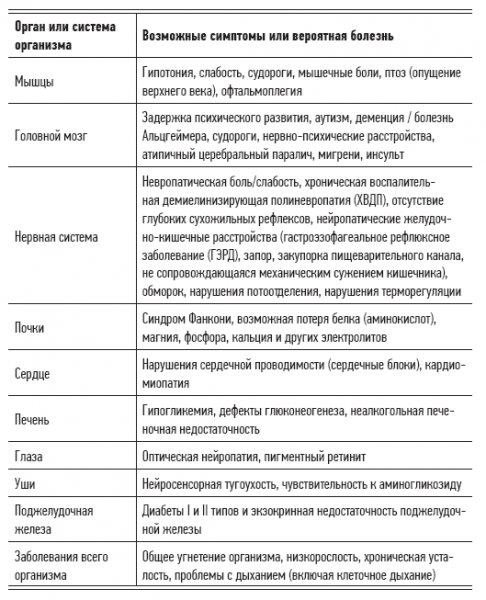
Tabelle 2.3. Angeborene Krankheiten, die durch mitochondriale Dysfunktion verursacht werden
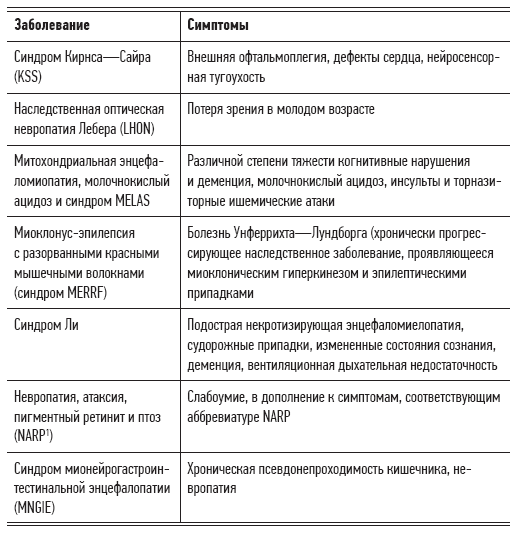
Natürlich benötigen einige Organe oder Gewebe mehr Energie als andere. Wenn die Energiebedürfnisse eines bestimmten Organs nicht vollständig gedeckt werden können, treten Symptome des mitochondrialen Syndroms auf. Zuallererst betreffen sie die Funktionen des Gehirns, des Nervensystems, der Muskeln, des Herzens, der Nieren und des endokrinen Systems, also aller Organe, die für ihre normale Funktion eine große Menge an Energie benötigen.
Erworbene Krankheiten, die durch mitochondriale Dysfunktion verursacht werden
Mit dem wachsenden Verständnis der mitochondrialen Funktion und Dysfunktion entwickeln wir eine umfangreiche Liste von Krankheiten, die auf mitochondrialer Dysfunktion beruhen, und klären die Mechanismen, die diesen Erkrankungen zugrunde liegen. Daten aus einigen aktuellen Studien zeigen, dass jeder 2500. Mensch an einem mitochondrialen Syndrom leidet. Wenn Sie jedoch die nachstehende Liste genauer betrachten, werden Sie zustimmen, dass es sehr wahrscheinlich ist, dass mitochondriale Erkrankungen (angeboren oder erworben) bald bei jedem fünften oder sogar zehnten Einwohner der westlichen Länder festgestellt werden.
- Typ-2-Diabetes
- Krebs
- Alzheimer-Krankheit
- Parkinson-Krankheit
- Bipolare affektive Störung
- Schizophrenie
- Alterung und Senilität
- Angststörung
- Nicht-alkoholische Steatohepatitis
- Herz-Kreislauf-Erkrankungen
- Sarkopenie (Verlust von Muskelmasse und -kraft)
- Belastungsintoleranz
- Müdigkeit, einschließlich chronisches Erschöpfungssyndrom, Fibromyalgie und myofasziale Schmerzen
Auf genetischer Ebene stehen damit sehr komplexe Prozesse im Zusammenhang. Die energetische Kraft einer bestimmten Person kann ermittelt werden, indem man angeborene Störungen ihrer mitochondrialen DNA untersucht. Doch dies ist lediglich der Ausgangspunkt. Im Laufe der Zeit sammeln sich im Körper erworbene Defekte der mtDNA an, und nachdem ein bestimmtes Organ eine gewisse Schwelle überschreitet, beginnt es, Schwierigkeiten zu haben oder wird anfällig für Degeneration (jede Organ hat eine eigene Toleranzgrenze, über die wir genauer sprechen werden).
Eine weitere Herausforderung besteht darin, dass jede Mitochondrie bis zu zehn Kopien von mtDNA enthält, und jede Zelle, jedes Gewebe und jedes Organ viele Mitochondrien hat. Daraus folgt, dass es in unserem Körper unzählige Fehler in den Kopien der mtDNA gibt. Die Dysfunktion eines bestimmten Organs beginnt, wenn der Anteil der defekten Mitochondrien darin einen bestimmten Schwellenwert übersteigt. Dieses Phänomen wird als Schwellenwert-Effekt bezeichnet. Jedes Organ und jedes Gewebe ist spezifischen Mutationen ausgesetzt und weist einen eigenen Mutationsschwellenwert, Energiebedarf und eine Widerstandsfähigkeit gegenüber freien Radikalen auf. Die Kombination dieser Faktoren bestimmt, wie genau ein lebendes System auf genetische Störungen reagiert.
Wenn nur 10 % der Mitochondrien defekt sind, können die 90 % der verbleibenden normalen Zellenergie-Generatoren die Dysfunktion ihrer "Kollegen" ausgleichen. Oder wenn zum Beispiel eine Mutation nicht sehr schwerwiegend ist, aber eine große Anzahl von Mitochondrien betrifft, kann die Zelle dennoch normal funktionieren.
Es gibt auch das Konzept der Segregation defekter Mitochondrien: Bei der Zellteilung werden die Mitochondrien zufällig zwischen zwei Tochterzellen verteilt. Eine dieser Zellen kann alle mutierten Mitochondrien erhalten, während die andere alle funktionstüchtigen "Kraftwerke" annimmt (natürlich sind auch Zwischenvarianten wahrscheinlicher). Zellen mit dysfunktionalen Mitochondrien sterben im Zuge der Apoptose, während gesunde weiterhin ihre Aufgaben erfüllen (eine der Erklärungen für das plötzliche und unerwartete Verschwinden des mitochondrialen Syndroms). Das Phänomen der Unterschiede in der DNA-Sequenz der Mitochondrien (oder Plastiden) im selben Organismus, oft sogar in derselben Zelle, wenn beispielsweise einige Mitochondrien eine pathologische Mutation aufweisen und andere nicht, wird als Heteroplasmie bezeichnet. Der Grad der Heteroplasmie variiert sogar innerhalb einer Familie. Darüber hinaus kann der Heteroplasmiestatus innerhalb eines Organismus je nach spezifischem Organ oder Zelle schwanken, was zu einem sehr breiten Spektrum an Manifestationen und Symptomen mitochondrialer Erkrankungen führt.
Im Körper eines wachsenden Embryos füllen sich die Organe und Gewebe mit mutierten Mitochondrien, während sich die Zellen teilen, und weisen unterschiedliche Energiebedarfe auf. Wenn in einem hohen Maße mutierte Mitochondrien in Zellen ansässig sind, die sich mit der Zeit in metabolisch aktive Strukturen verwandeln (wie zum Beispiel im Gehirn oder Herzen), kann es für den entsprechenden Organismus zu Problemen mit der Lebensqualität kommen (oder er ist überhaupt nicht lebensfähig). Auf der anderen Seite, wenn sich eine Masse dysfunktionaler Mitochondrien vor allem in Zellen ansammelt, die einen geringen Stoffwechsel haben (zum Beispiel in Hautzellen, die regelmäßig ersetzt werden), kann der Träger solcher Mitochondrien nie von seiner genetischen Veranlagung zum mitochondrialen Syndrom erfahren. In dem oben genannten Episoden aus "Der Bachelor" schien eines der Mädchen mit einer mitochondrialen Erkrankung vollkommen normal zu sein, während eine andere offensichtlich unter einer ernsthaften Erkrankung litt.
Einige mitochondriale Mutationen entwickeln sich spontan mit dem Alter durch die Bildung von freien Radikalen im Verlauf des normalen Metabolismus. Was dann geschieht, hängt von einer Reihe von Faktoren ab. Wenn beispielsweise eine mit dysfunktionalen Mitochondrien gefüllte Zelle sich schnell teilt, wie es Stammzellen tun, die für die Geweberegeneration verantwortlich sind, werden die defekten Energieerzeuger aktiv expandieren. Wenn hingegen eine geschwächte Zelle sich nicht mehr teilt (nehmen wir an, es handelt sich um ein Neuron), bleiben die Mutationen nur auf diese Zelle beschränkt, was jedoch nicht die Möglichkeit einer erfolgreichen zufälligen Mutation ausschließt. Daher erklärt sich die Komplexität der genetischen Basis des mitochondrialen Syndroms durch die Tatsache, dass die Erschöpfung der bioenergetischen Ressourcen des Körpers, die durch mitochondrialen Mutationen verursacht wird, sich in einer Vielzahl unterschiedlicher und komplexer Krankheiten und Symptome zeigt.
Wir müssen auch bedenken, dass es viele Gene außerhalb der mtDNA gibt, die für das normale Funktionieren der Mitochondrien verantwortlich sind. Wenn eine Mutation Gene betrifft, die RNA kodieren, können die Folgen in der Regel sehr ernst sein. In Fällen, in denen ein Kind bei seiner Empfängnis einen mutierten Transkriptionsfaktor der Mitochondrien von einem der Elternteile erbt (erinnern wir uns, dass Transkriptionsfaktoren Proteine sind, die den Prozess der Synthese von mRNA auf der DNA-Matrix steuern, indem sie sich an spezifische DNA-Abschnitte binden), sind alle Mitochondrien des Organismus dem pathogenetischen Einfluss ausgesetzt. Wenn die Mutation jedoch nur spezifische Transkriptionsfaktoren betrifft, die nur in bestimmten Organen oder Geweben oder als Reaktion auf die Ausschüttung eines bestimmten Hormons aktiviert werden, wird der entsprechende pathogenetische Effekt ausschließlich lokal sein.
Die Vielzahl von mitochondrialen Erkrankungen und deren Ausprägungen stellt eine ernsthafte Herausforderung für Mediziner dar, sowohl im theoretischen als auch im praktischen Bereich, einschließlich der tatsächlichen Unmöglichkeit, den Verlauf des mitochondrialen Syndroms vorherzusagen. Es gibt so viele mitochondriale Erkrankungen, dass es schwierig ist, ihnen einfach Namen zu geben, und viele davon sind noch nicht entdeckt. Sogar eine Reihe bekannter degenerativer Krankheiten (Krankheiten des Herz-Kreislauf-Systems, Krebserkrankungen, verschiedene Formen von Demenz usw.) werden von der modernen Wissenschaft als Folge von Mitochondriendysfunktion betrachtet.
Es ist wichtig zu erkennen, dass, obwohl es keine vollständige Heilung für mitochondriale Erkrankungen gibt, viele Menschen mit diesen Beschwerden (insbesondere wenn es sich um eine leichte oder mittelschwere Form der Krankheit handelt) ein langes und erfülltes Leben führen können. Dafür ist jedoch ein systematisches Vorgehen erforderlich, indem wir das Wissen, das uns zur Verfügung steht, nutzen.
Über den Autor
Li Nou — ein lizenzierter Naturheilpraktiker aus Kanada, mehrfach ausgezeichnet. Kollegen kennen ihn als vorausschauenden Unternehmer, Strategen und Arzt. Lee hatte Positionen als medizinischer Berater, wissenschaftlicher Experte und Leiter für Forschung und Entwicklung in großen Organisationen inne. Neben seiner wissenschaftlichen Tätigkeit in seiner Firma berät er auch im Bereich natürlicher Gesundheitsprodukte und Nahrungsergänzungsmittel und ist Mitglied des editorial advisory board des Magazins Alive – des meistgelesenen Gesundheitsmagazins in Kanada. Er nennt die Region Greater Toronto sein Zuhause, wo er mit seiner Frau und ihren beiden Söhnen lebt, und hat ein besonderes Interesse an der Stärkung der natürlichen Gesundheit und dem Umweltschutz.
» Weitere Informationen zu dem Buch finden Sie auf
»
»
Für Habr-Besucher gilt ein Rabatt von 25 % mit dem Code — Mitochondrien
Nach Erhalt der Zahlung für die gedruckte Version des Buches wird das E-Book per E-Mail versendet.
Quelle: habr.com
