


Cara membaca artikel ini: Saya minta maaf karena teksnya panjang dan kacau. Untuk menghemat waktu Anda, saya memulai setiap bab dengan pendahuluan “Apa yang Saya Pelajari”, yang merangkum inti bab ini dalam satu atau dua kalimat.
“Tunjukkan saja padaku solusinya!” Jika Anda hanya ingin melihat dari mana saya berasal, lewati saja ke bab “Menjadi Lebih Inventif”, namun menurut saya akan lebih menarik dan berguna untuk membaca tentang kegagalan.
Saya baru-baru ini ditugaskan untuk menyiapkan proses untuk memproses rangkaian DNA mentah dalam jumlah besar (secara teknis merupakan chip SNP). Kebutuhannya adalah memperoleh data dengan cepat tentang lokasi genetik tertentu (disebut SNP) untuk pemodelan selanjutnya dan tugas lainnya. Dengan menggunakan R dan AWK, saya dapat membersihkan dan mengatur data dengan cara alami, sehingga mempercepat pemrosesan kueri. Ini tidak mudah bagi saya dan memerlukan banyak pengulangan. Artikel ini akan membantu Anda menghindari beberapa kesalahan saya dan menunjukkan kepada Anda apa yang akhirnya saya lakukan.
Pertama, beberapa penjelasan pendahuluan.
Data
Pusat pemrosesan informasi genetik universitas kami memberi kami data dalam bentuk TSV 25 TB. Saya menerimanya dibagi menjadi 5 paket terkompresi Gzip, masing-masing berisi sekitar 240 file berukuran empat gigabyte. Setiap baris berisi data untuk satu SNP dari satu individu. Secara total, data ~2,5 juta SNP dan ~60 ribu orang dikirimkan. Selain informasi SNP, file tersebut berisi banyak kolom dengan angka yang mencerminkan berbagai karakteristik, seperti intensitas baca, frekuensi alel yang berbeda, dll. Total ada sekitar 30 kolom dengan nilai unik.
target
Seperti halnya proyek pengelolaan data lainnya, hal terpenting adalah menentukan bagaimana data akan digunakan. Pada kasus ini kami sebagian besar akan memilih model dan alur kerja untuk SNP berdasarkan SNP. Artinya, kita hanya membutuhkan data pada satu SNP dalam satu waktu. Saya harus belajar cara mengambil semua catatan yang terkait dengan salah satu dari 2,5 juta SNP dengan mudah, cepat, dan semurah mungkin.
Bagaimana tidak melakukan ini
Mengutip ungkapan klise yang cocok:
Saya tidak gagal ribuan kali, saya hanya menemukan seribu cara untuk menghindari penguraian banyak data dalam format yang ramah kueri.
Percobaan pertama
Apa yang telah saya pelajari: Tidak ada cara murah untuk mengurai 25 TB sekaligus.
Setelah mengikuti kursus “Metode Tingkat Lanjut untuk Pemrosesan Data Besar” di Universitas Vanderbilt, saya yakin bahwa triknya ada di kantong. Mungkin diperlukan waktu satu atau dua jam untuk menyiapkan server Hive agar menjalankan semua data dan melaporkan hasilnya. Karena data kami disimpan di AWS S3, saya menggunakan layanan ini , yang memungkinkan Anda menerapkan kueri Hive SQL ke data S3. Anda tidak perlu menyiapkan/membangun cluster Hive, dan Anda juga hanya membayar untuk data yang Anda cari.
Setelah saya menunjukkan kepada Athena data saya dan formatnya, saya menjalankan beberapa tes dengan pertanyaan seperti ini:
select * from intensityData limit 10;Dan dengan cepat menerima hasil yang terstruktur dengan baik. Siap.
Sampai kami mencoba menggunakan data tersebut dalam pekerjaan kami...
Saya diminta mengeluarkan semua informasi SNP untuk menguji modelnya. Saya menjalankan kueri:
select * from intensityData
where snp = 'rs123456';...dan mulai menunggu. Setelah delapan menit dan lebih dari 4 TB data yang diminta, saya menerima hasilnya. Athena mengenakan biaya berdasarkan volume data yang ditemukan, $5 per terabyte. Jadi permintaan tunggal ini membutuhkan biaya $20 dan waktu tunggu delapan menit. Untuk menjalankan model pada semua data, kami harus menunggu 38 tahun dan membayar $50 juta. Tentu saja, ini tidak cocok untuk kami.
Itu perlu untuk menggunakan Parket...
Apa yang telah saya pelajari: Hati-hati dengan ukuran file Parket Anda dan organisasinya.
Saya pertama kali mencoba memperbaiki situasi dengan mengonversi semua TSV menjadi . Mereka nyaman untuk bekerja dengan kumpulan data besar karena informasi di dalamnya disimpan dalam bentuk kolom: setiap kolom terletak di segmen memori/disknya sendiri, berbeda dengan file teks, yang barisnya berisi elemen setiap kolom. Dan jika Anda perlu menemukan sesuatu, baca saja kolom yang diperlukan. Selain itu, setiap file menyimpan rentang nilai dalam sebuah kolom, jadi jika nilai yang Anda cari tidak berada dalam rentang kolom tersebut, Spark tidak akan membuang waktu untuk memindai seluruh file.
Saya menjalankan tugas sederhana untuk mengonversi TSV kami ke Parket dan memasukkan file baru ke Athena. Butuh waktu sekitar 5 jam. Namun saat saya menjalankan permintaan, dibutuhkan waktu yang hampir sama dan biaya yang lebih sedikit untuk menyelesaikannya. Faktanya adalah Spark, yang mencoba mengoptimalkan tugasnya, cukup membongkar satu potongan TSV dan memasukkannya ke dalam potongan Parketnya sendiri. Dan karena setiap potongan cukup besar untuk memuat seluruh catatan banyak orang, setiap file berisi semua SNP, jadi Spark harus membuka semua file untuk mengekstrak informasi yang diperlukan.
Menariknya, jenis kompresi default Parket (dan direkomendasikan), tajam, tidak dapat dipisahkan. Oleh karena itu, setiap pelaksana terjebak pada tugas membongkar dan mengunduh kumpulan data 3,5 GB secara lengkap.
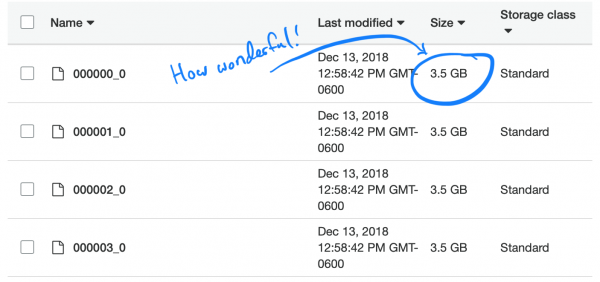
Mari kita pahami masalahnya
Apa yang telah saya pelajari: Penyortiran itu sulit, apalagi jika datanya tersebar.
Tampaknya bagi saya sekarang saya memahami inti permasalahannya. Saya hanya perlu mengurutkan data berdasarkan kolom SNP, bukan berdasarkan orang. Kemudian beberapa SNP akan disimpan dalam potongan data terpisah, dan kemudian fungsi "pintar" Parket "terbuka hanya jika nilainya berada dalam kisaran" akan menunjukkan dirinya dengan segala kejayaannya. Sayangnya, memilah miliaran baris yang tersebar di sebuah cluster terbukti menjadi tugas yang sulit.
Saya mengambil kelas algoritme di perguruan tinggi: “Ugh, tidak ada yang peduli dengan kompleksitas komputasi dari semua algoritme pengurutan ini”
Saya mencoba mengurutkan kolom dalam 20TB tabel: “Mengapa ini memakan waktu lama?” perjuangan.
— Nick Strayer (@NicholasStrayer)
AWS jelas tidak ingin memberikan pengembalian dana karena alasan "Saya adalah siswa yang perhatiannya terganggu". Setelah saya menjalankan penyortiran di Amazon Glue, penyortiran berjalan selama 2 hari dan mogok.
Bagaimana dengan mempartisi?
Apa yang telah saya pelajari: Partisi di Spark harus seimbang.
Kemudian saya mendapat ide untuk mempartisi data dalam kromosom. Ada 23 di antaranya (dan masih banyak lagi jika Anda memperhitungkan DNA mitokondria dan wilayah yang belum dipetakan).
Ini akan memungkinkan Anda membagi data menjadi beberapa bagian yang lebih kecil. Jika Anda menambahkan hanya satu baris ke fungsi ekspor Spark di skrip Glue partition_by = "chr", maka data harus dibagi menjadi beberapa kelompok.
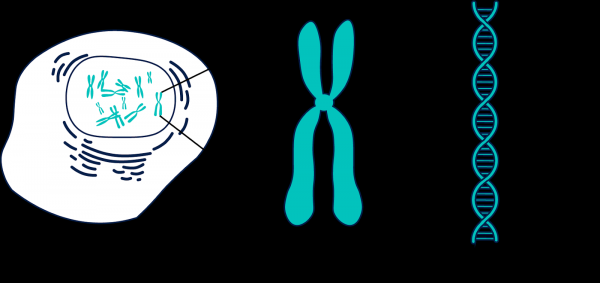
Genom terdiri dari banyak fragmen yang disebut kromosom.
Sayangnya, itu tidak berhasil. Kromosom memiliki ukuran berbeda, yang berarti jumlah informasi berbeda. Artinya, tugas yang dikirim Spark ke pekerja tidak seimbang dan diselesaikan dengan lambat karena beberapa node selesai lebih awal dan menganggur. Namun, tugas telah selesai. Namun ketika meminta satu SNP, ketidakseimbangan tersebut kembali menimbulkan masalah. Biaya pemrosesan SNP pada kromosom yang lebih besar (yaitu, tempat kita ingin mendapatkan data) hanya berkurang sekitar 10 kali lipat. Banyak, tapi tidak cukup.
Bagaimana jika kita membaginya menjadi beberapa bagian yang lebih kecil?
Apa yang telah saya pelajari: Jangan pernah mencoba melakukan 2,5 juta partisi sama sekali.
Saya memutuskan untuk berusaha sekuat tenaga dan mempartisi setiap SNP. Ini memastikan bahwa partisi memiliki ukuran yang sama. ITU ADALAH IDE YANG BURUK. Saya menggunakan Lem dan menambahkan garis polos partition_by = 'snp'. Tugas dimulai dan mulai dilaksanakan. Sehari kemudian saya memeriksa dan melihat bahwa masih belum ada yang ditulis ke S3, jadi saya menghentikan tugas tersebut. Sepertinya Glue sedang menulis file perantara ke lokasi tersembunyi di S3, banyak file, mungkin beberapa juta. Akibatnya, kesalahan saya merugikan lebih dari seribu dolar dan tidak menyenangkan mentor saya.
Partisi + penyortiran
Apa yang telah saya pelajari: Penyortiran masih sulit, begitu pula penyetelan Spark.
Upaya terakhir saya dalam mempartisi melibatkan saya mempartisi kromosom dan kemudian mengurutkan setiap partisi. Secara teori, hal ini akan mempercepat setiap kueri karena data SNP yang diinginkan harus berada dalam beberapa potongan Parket dalam rentang tertentu. Sayangnya, menyortir data yang dipartisi ternyata menjadi tugas yang sulit. Hasilnya, saya beralih ke EMR untuk klaster khusus dan menggunakan delapan instans canggih (C5.4xl) dan Sparklyr untuk menciptakan alur kerja yang lebih fleksibel...
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)...namun, tugas tersebut masih belum selesai. Saya mengonfigurasinya dengan cara yang berbeda: meningkatkan alokasi memori untuk setiap pelaksana kueri, menggunakan node dengan jumlah memori yang besar, menggunakan variabel siaran (variabel penyiaran), tetapi setiap kali ini menjadi setengah-setengah, dan secara bertahap para pelaksana mulai untuk gagal sampai semuanya berhenti.
Pembaruan: begitulah dimulai.
— Nick Strayer (@NicholasStrayer)
Saya menjadi lebih kreatif
Apa yang telah saya pelajari: Terkadang data khusus memerlukan solusi khusus.
Setiap SNP memiliki nilai posisi. Ini adalah angka yang sesuai dengan jumlah basa di sepanjang kromosomnya. Ini adalah cara yang bagus dan alami untuk mengatur data kami. Awalnya saya ingin mempartisi berdasarkan wilayah setiap kromosom. Misalnya posisi 1 - 2000, 2001 - 4000, dst. Namun masalahnya adalah SNP tidak terdistribusi secara merata di seluruh kromosom, sehingga ukuran kelompoknya akan sangat bervariasi.
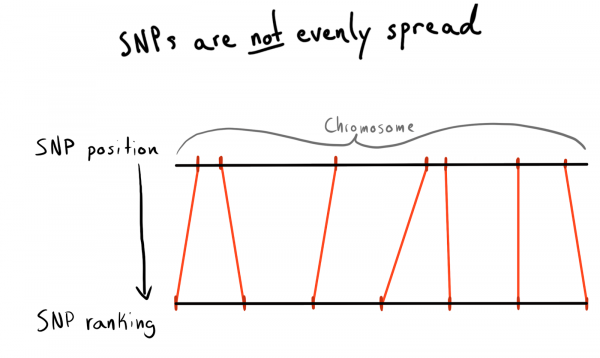
Hasilnya, saya sampai pada pengelompokan posisi ke dalam kategori (peringkat). Dengan menggunakan data yang sudah diunduh, saya menjalankan permintaan untuk mendapatkan daftar SNP unik, posisi dan kromosomnya. Kemudian saya mengurutkan data dalam setiap kromosom dan mengumpulkan SNP ke dalam kelompok (bin) dengan ukuran tertentu. Katakanlah masing-masing 1000 SNP. Ini memberi saya hubungan SNP-ke-kelompok-per-kromosom.
Pada akhirnya saya membuat kelompok (bin) dari 75 SNP, alasannya akan dijelaskan di bawah ini.
snp_to_bin <- unique_snps %>%
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()Coba dulu dengan Spark
Apa yang telah saya pelajari: Agregasi Spark cepat, namun partisi masih mahal.
Saya ingin membaca bingkai data kecil (2,5 juta baris) ini ke dalam Spark, menggabungkannya dengan data mentah, dan kemudian mempartisinya dengan kolom yang baru ditambahkan bin.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
)
saya menggunakan sdf_broadcast(), jadi Spark mengetahui bahwa ia harus mengirimkan bingkai data ke semua node. Ini berguna jika data berukuran kecil dan diperlukan untuk semua tugas. Jika tidak, Spark akan mencoba menjadi cerdas dan mendistribusikan data sesuai kebutuhan, yang dapat menyebabkan perlambatan.
Dan lagi, ide saya tidak berhasil: tugas-tugas tersebut berhasil selama beberapa waktu, menyelesaikan penyatuan, dan kemudian, seperti para pelaksana yang diluncurkan dengan mempartisi, mereka mulai gagal.
Menambahkan AWK
Apa yang telah saya pelajari: Jangan tidur ketika Anda sedang diajari dasar-dasarnya. Tentunya seseorang telah memecahkan masalah Anda pada tahun 1980an.
Hingga saat ini, alasan semua kegagalan saya dengan Spark adalah campur aduknya data di cluster. Mungkin situasinya dapat diperbaiki dengan pra-perawatan. Saya memutuskan untuk mencoba membagi data teks mentah menjadi kolom-kolom kromosom, jadi saya berharap dapat memberikan Spark data yang “dipartisi sebelumnya”.
Saya mencari di StackOverflow tentang cara membagi berdasarkan nilai kolom dan menemukannya Dengan AWK Anda dapat membagi file teks berdasarkan nilai kolom dengan menuliskannya dalam skrip daripada mengirimkan hasilnya ke stdout.
Saya menulis skrip Bash untuk mencobanya. Mengunduh salah satu TSV yang dikemas, lalu membongkarnya menggunakan gzip dan dikirim ke awk.
gzip -dc path/to/chunk/file.gz |
awk -F 't'
'{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'Itu berhasil!
Mengisi inti
Apa yang telah saya pelajari: gnu parallel - itu adalah hal yang ajaib, semua orang harus menggunakannya.
Pemisahannya cukup lambat dan ketika saya mulai htopuntuk memeriksa penggunaan instance EC2 yang kuat (dan mahal), ternyata saya hanya menggunakan satu inti dan memori sekitar 200 MB. Untuk mengatasi masalah ini dan tidak kehilangan banyak uang, kami harus memikirkan cara untuk memparalelkan pekerjaan. Untungnya, dalam sebuah buku yang benar-benar menakjubkan Saya menemukan bab oleh Jeron Janssens tentang paralelisasi. Dari situ saya belajar tentang gnu parallel, metode yang sangat fleksibel untuk mengimplementasikan multithreading di Unix.

Ketika saya memulai partisi menggunakan proses baru, semuanya baik-baik saja, tetapi masih ada hambatan - pengunduhan objek S3 ke disk tidak terlalu cepat dan tidak sepenuhnya diparalelkan. Untuk memperbaikinya, saya melakukan ini:
- Saya menemukan bahwa tahap pengunduhan S3 dapat diimplementasikan secara langsung dalam pipeline, sepenuhnya menghilangkan penyimpanan perantara pada disk. Ini berarti saya dapat menghindari penulisan data mentah ke disk dan menggunakan penyimpanan yang lebih kecil, sehingga lebih murah, di AWS.
- tim
aws configure set default.s3.max_concurrent_requests 50sangat meningkatkan jumlah thread yang digunakan AWS CLI (secara default ada 10). - Saya beralih ke instans EC2 yang dioptimalkan untuk kecepatan jaringan, dengan huruf n di namanya. Saya menemukan bahwa hilangnya daya pemrosesan saat menggunakan n-instance lebih dari sekadar dikompensasi oleh peningkatan kecepatan pemuatan. Untuk sebagian besar tugas saya menggunakan c5n.4xl.
- Berubah
gzippada , ini adalah alat gzip yang dapat melakukan hal-hal keren untuk memparalelkan tugas dekompresi file yang awalnya tidak diparalelkan (ini paling tidak membantu).
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe
"awk -F 't' '{print $1",..."$30">"chunked/{#}_chr"$15".csv"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneLangkah-langkah ini digabungkan satu sama lain untuk membuat semuanya bekerja dengan sangat cepat. Dengan meningkatkan kecepatan pengunduhan dan menghilangkan penulisan disk, kini saya dapat memproses paket 5 terabyte hanya dalam beberapa jam.
Tidak ada yang lebih menyenangkan daripada melihat semua inti yang Anda bayar di AWS digunakan. Berkat gnu-parallel saya dapat mengekstrak dan membagi csv 19gig secepat saya dapat mengunduhnya. Aku bahkan tidak bisa mendapatkan percikan untuk menjalankan ini.
— Nick Strayer (@NicholasStrayer)
Tweet ini seharusnya menyebutkan 'TSV'. Sayang.
Menggunakan data yang baru diurai
Apa yang telah saya pelajari: Spark menyukai data yang tidak terkompresi dan tidak suka menggabungkan partisi.
Sekarang datanya ada di S3 dalam format yang belum dibongkar (baca: dibagikan) dan setengah dipesan, dan saya bisa kembali ke Spark lagi. Sebuah kejutan menanti saya: Saya kembali gagal mencapai apa yang saya inginkan! Sangat sulit untuk memberi tahu Spark dengan tepat bagaimana data dipartisi. Dan ketika saya melakukan ini, ternyata partisinya terlalu banyak (95 ribu), dan ketika saya menggunakannya coalesce mengurangi jumlahnya hingga batas wajar, ini menghancurkan partisi saya. Saya yakin ini dapat diperbaiki, tetapi setelah beberapa hari mencari, saya tidak dapat menemukan solusi. Saya akhirnya menyelesaikan semua tugas di Spark, meskipun butuh waktu cukup lama dan file Parket saya yang terbagi tidak terlalu kecil (~200 KB). Namun, datanya ada di tempat yang dibutuhkan.
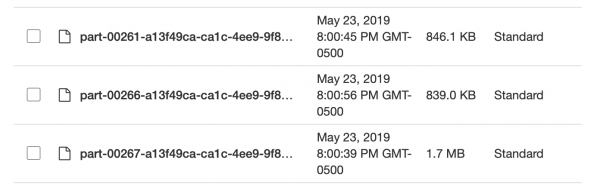
Terlalu kecil dan tidak rata, luar biasa!
Menguji kueri Spark lokal
Apa yang telah saya pelajari: Spark memiliki terlalu banyak overhead saat memecahkan masalah sederhana.
Dengan mengunduh data dalam format yang cerdas, saya dapat menguji kecepatannya. Siapkan skrip R untuk menjalankan server Spark lokal, lalu muat bingkai data Spark dari penyimpanan grup Parket (bin) yang ditentukan. Saya mencoba memuat semua data tetapi tidak dapat membuat Sparklyr mengenali partisi tersebut.
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Start a timer
start_time <- Sys.time()
# Load the desired bin into Spark
intensity_data <- sc %>%
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Subset bin to snp and then collect to local
test_subset <- intensity_data %>%
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)Eksekusinya memakan waktu 29,415 detik. Jauh lebih baik, tetapi tidak terlalu bagus untuk pengujian massal apa pun. Selain itu, saya tidak dapat mempercepat proses caching karena ketika saya mencoba melakukan cache bingkai data di memori, Spark selalu mogok, bahkan ketika saya mengalokasikan lebih dari 50 GB memori ke kumpulan data yang beratnya kurang dari 15.
Kembali ke AWK
Apa yang telah saya pelajari: Array asosiatif di AWK sangat efisien.
Saya menyadari bahwa saya dapat mencapai kecepatan yang lebih tinggi. Saya ingat itu dengan luar biasa Saya membaca tentang fitur keren yang disebut “" Pada dasarnya, ini adalah pasangan nilai kunci, yang karena alasan tertentu disebut berbeda di AWK, dan oleh karena itu saya tidak terlalu memikirkannya. ingat bahwa istilah “array asosiatif” jauh lebih tua daripada istilah “pasangan nilai kunci”. Bahkan jika kamu , Anda tidak akan melihat istilah ini di sana, tetapi Anda akan menemukan array asosiatif! Selain itu, “pasangan nilai kunci” paling sering dikaitkan dengan database, jadi lebih masuk akal untuk membandingkannya dengan peta hash. Saya menyadari bahwa saya dapat menggunakan array asosiatif ini untuk mengaitkan SNP saya dengan tabel bin dan data mentah tanpa menggunakan Spark.
Untuk melakukan ini, dalam skrip AWK saya menggunakan blok BEGIN. Ini adalah bagian kode yang dieksekusi sebelum baris data pertama diteruskan ke bagian utama skrip.
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline < "snp_to_bin.csv") {bin[$1] = $2}
}
{
print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
}
Tim while(getline...) memuat semua baris dari grup CSV (bin), atur kolom pertama (nama SNP) sebagai kunci untuk array asosiatif bin dan nilai kedua (grup) sebagai nilainya. Lalu di blok { }, yang dijalankan pada semua baris file utama, setiap baris dikirim ke file keluaran, yang menerima nama unik tergantung pada grupnya (bin): ..._bin_"bin[$1]"_....
Variabel batch_num и chunk_id cocok dengan data yang disediakan oleh pipeline, menghindari kondisi balapan, dan setiap thread eksekusi berjalan parallel, menulis ke file uniknya sendiri.
Karena saya menyebarkan semua data mentah ke dalam folder pada kromosom yang tersisa dari percobaan saya sebelumnya dengan AWK, sekarang saya dapat menulis skrip Bash lain untuk memproses satu kromosom pada satu waktu dan mengirim data yang dipartisi lebih dalam ke S3.
DESIRED_CHR='13'
# Download chromosome data from s3 and split into bins
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'reading {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=""$DESIRED_CHR"" -v chunk="{}" -f split_on_chr_bin.awk"
# Combine all the parallel process chunks to single files and upload to rds using R
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/*
Script memiliki dua bagian parallel.
Pada bagian pertama, data dibaca dari semua file yang berisi informasi tentang kromosom yang diinginkan, kemudian data ini didistribusikan ke seluruh thread, yang mendistribusikan file ke dalam grup (bin) yang sesuai. Untuk menghindari kondisi balapan ketika beberapa thread menulis ke file yang sama, AWK meneruskan nama file untuk menulis data ke tempat berbeda, misalnya. chr_10_bin_52_batch_2_aa.csv. Akibatnya, banyak file kecil dibuat di disk (untuk ini saya menggunakan volume EBS terabyte).
Konveyor dari bagian kedua parallel menelusuri grup (bin) dan menggabungkan file individualnya ke dalam CSV umum c catdan kemudian mengirimkannya untuk diekspor.
Menyiarkan di R?
Apa yang telah saya pelajari: Anda dapat menghubungi stdin и stdout dari skrip R, dan karena itu menggunakannya dalam pipeline.
Anda mungkin memperhatikan baris ini di skrip Bash Anda: ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... Ini menerjemahkan semua file grup gabungan (bin) ke dalam skrip R di bawah. {} adalah teknik khusus parallel, yang memasukkan data apa pun yang dikirimkannya ke aliran tertentu langsung ke dalam perintah itu sendiri. Pilihan {#} memberikan ID thread unik, dan {%} mewakili nomor slot pekerjaan (berulang, tetapi tidak pernah secara bersamaan). Daftar semua opsi dapat ditemukan di
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
)
Ketika sebuah variabel file("stdin") ditransmisikan ke readr::read_csv, data yang diterjemahkan ke dalam skrip R dimuat ke dalam bingkai, yang kemudian berbentuk .rds-mengajukan menggunakan aws.s3 ditulis langsung ke S3.
RDS adalah semacam Parket versi junior, tanpa embel-embel penyimpanan speaker.
Setelah menyelesaikan skrip Bash saya mendapat bundel .rds-file terletak di S3, yang memungkinkan saya menggunakan kompresi yang efisien dan tipe bawaan.
Meski menggunakan rem R, semuanya bekerja sangat cepat. Tidak mengherankan, bagian R yang membaca dan menulis data sangat dioptimalkan. Setelah pengujian pada satu kromosom berukuran sedang, pekerjaan diselesaikan pada instance C5n.4xl dalam waktu sekitar dua jam.
Keterbatasan S3
Apa yang telah saya pelajari: Berkat penerapan jalur cerdas, S3 dapat menangani banyak file.
Saya khawatir apakah S3 mampu menangani banyak file yang ditransfer ke dalamnya. Saya dapat membuat nama file masuk akal, tetapi bagaimana S3 mencarinya?

Folder di S3 hanya untuk pajangan saja, nyatanya sistem tidak tertarik dengan simbolnya /.
Tampaknya S3 mewakili jalur ke file tertentu sebagai kunci sederhana dalam semacam tabel hash atau database berbasis dokumen. Sebuah keranjang dapat dianggap sebagai sebuah tabel, dan file dapat dianggap sebagai catatan dalam tabel tersebut.
Karena kecepatan dan efisiensi penting untuk menghasilkan keuntungan di Amazon, tidak mengherankan jika sistem jalur kunci sebagai file ini sangat dioptimalkan. Saya mencoba mencari keseimbangan: sehingga saya tidak perlu membuat banyak permintaan get, tetapi permintaan tersebut dieksekusi dengan cepat. Ternyata yang terbaik adalah membuat sekitar 20 ribu file bin. Saya rasa jika kita terus melakukan optimasi, kita bisa mencapai peningkatan kecepatan (misalnya membuat bucket khusus hanya untuk data, sehingga mengurangi ukuran tabel pencarian). Tapi tidak ada waktu atau uang untuk eksperimen lebih lanjut.
Bagaimana dengan kompatibilitas silang?
Apa yang Saya Pelajari: Penyebab nomor satu dari waktu yang terbuang adalah mengoptimalkan metode penyimpanan Anda sebelum waktunya.
Pada titik ini, sangat penting untuk bertanya pada diri sendiri: “Mengapa menggunakan format file berpemilik?” Alasannya terletak pada kecepatan pemuatan (file CSV yang di-gzip membutuhkan waktu 7 kali lebih lama untuk dimuat) dan kompatibilitas dengan alur kerja kami. Saya mungkin mempertimbangkan kembali apakah R dapat dengan mudah memuat file Parket (atau Panah) tanpa memuat Spark. Semua orang di lab kami menggunakan R, dan jika saya perlu mengonversi data ke format lain, saya masih memiliki data teks asli, jadi saya bisa menjalankan pipeline lagi.
pembagian kerja
Apa yang telah saya pelajari: Jangan mencoba mengoptimalkan pekerjaan secara manual, biarkan komputer yang melakukannya.
Saya telah men-debug alur kerja pada satu kromosom, sekarang saya perlu memproses semua data lainnya.
Saya ingin meningkatkan beberapa instans EC2 untuk konversi, tetapi pada saat yang sama saya takut mendapatkan beban yang sangat tidak seimbang di berbagai pekerjaan pemrosesan (seperti Spark yang mengalami partisi yang tidak seimbang). Selain itu, saya tidak tertarik untuk menaikkan satu instance per kromosom, karena untuk akun AWS ada batas default 10 instance.
Kemudian saya memutuskan untuk menulis skrip di R untuk mengoptimalkan pekerjaan pemrosesan.
Pertama, saya meminta S3 menghitung berapa banyak ruang penyimpanan yang ditempati setiap kromosom.
library(aws.s3)
library(tidyverse)
chr_sizes <- get_bucket_df(
bucket = '...', prefix = '...', max = Inf
) %>%
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Extract chromosome from the file name
chr = str_extract(Key, 'chr.{1,4}.csv') %>%
str_remove_all('chr|.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Divide to get value in GB
# A tibble: 27 x 2
chr total_size
<chr> <dbl>
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … with 17 more rows
Lalu saya menulis fungsi yang mengambil ukuran total, mengacak urutan kromosom, membaginya menjadi beberapa kelompok num_jobs dan memberi tahu Anda betapa berbedanya ukuran semua pekerjaan pemrosesan.
num_jobs <- 7
# How big would each job be if perfectly split?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job <- function(i){
chr_sizes %>%
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)
# A tibble: 1 x 2
sd data
<dbl> <list>
1 153. <tibble [7 × 3]>Kemudian saya melakukan seribu pengacakan menggunakan purrr dan memilih yang terbaik.
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1)
Jadi saya mendapatkan serangkaian tugas yang ukurannya sangat mirip. Maka yang tersisa hanyalah membungkus skrip Bash saya sebelumnya dalam satu lingkaran besar for. Pengoptimalan ini membutuhkan waktu sekitar 10 menit untuk ditulis. Dan ini jauh lebih sedikit daripada yang saya habiskan untuk membuat tugas secara manual jika tugas tersebut tidak seimbang. Oleh karena itu, menurut saya optimasi awal ini benar.
for DESIRED_CHR in "16" "9" "7" "21" "MT"
do
# Code for processing a single chromosome
fiPada akhirnya saya menambahkan perintah shutdown:
sudo shutdown -h now
... dan semuanya berhasil! Dengan menggunakan AWS CLI, saya memunculkan instance menggunakan opsi user_data memberi mereka skrip Bash tentang tugas mereka untuk diproses. Mereka berjalan dan mati secara otomatis, jadi saya tidak membayar untuk tenaga pemrosesan ekstra.
aws ec2 run-instances ...
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]"
--user-data file://<<job_script_loc>>Ayo berkemas!
Apa yang telah saya pelajari: API harus sederhana demi kemudahan dan fleksibilitas penggunaan.
Akhirnya saya mendapatkan datanya pada tempat dan bentuk yang tepat. Tinggal menyederhanakan proses penggunaan data semaksimal mungkin agar memudahkan rekan-rekan saya. Saya ingin membuat API sederhana untuk membuat permintaan. Jika di kemudian hari saya memutuskan untuk beralih dari .rds ke file Parket, maka ini seharusnya menjadi masalah bagi saya, bukan bagi rekan-rekan saya. Untuk ini saya memutuskan untuk membuat paket R internal.
Bangun dan dokumentasikan paket yang sangat sederhana yang hanya berisi beberapa fungsi akses data yang diatur berdasarkan suatu fungsi get_snp. Saya juga membuat website untuk rekan-rekan saya , sehingga mereka dapat dengan mudah melihat contoh dan dokumentasi.
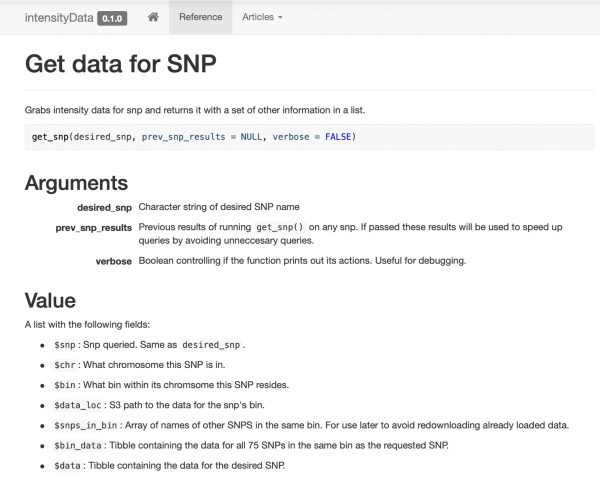
Penyimpanan cache yang cerdas
Apa yang telah saya pelajari: Jika data Anda sudah dipersiapkan dengan baik, caching akan mudah!
Karena salah satu alur kerja utama menerapkan model analisis yang sama ke paket SNP, saya memutuskan untuk menggunakan binning untuk keuntungan saya. Saat mengirimkan data melalui SNP, semua informasi dari grup (bin) dilampirkan ke objek yang dikembalikan. Artinya, kueri lama (secara teori) dapat mempercepat pemrosesan kueri baru.
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
...
Saat membuat paket, saya menjalankan banyak tolok ukur untuk membandingkan kecepatan saat menggunakan metode yang berbeda. Saya anjurkan untuk tidak mengabaikan hal ini, karena terkadang hasilnya tidak terduga. Misalnya, dplyr::filter jauh lebih cepat daripada mengambil baris menggunakan pemfilteran berbasis pengindeksan, dan mengambil satu kolom dari bingkai data yang difilter jauh lebih cepat daripada menggunakan sintaks pengindeksan.
Harap dicatat bahwa objeknya prev_snp_results berisi kuncinya snps_in_bin. Ini adalah array dari semua SNP unik dalam grup (bin), memungkinkan Anda memeriksa dengan cepat apakah Anda sudah memiliki data dari kueri sebelumnya. Ini juga memudahkan untuk mengulang semua SNP dalam grup (bin) dengan kode ini:
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}Temuan
Sekarang kami dapat (dan mulai serius) menjalankan model dan skenario yang sebelumnya tidak dapat kami akses. Hal terbaiknya adalah rekan lab saya tidak perlu memikirkan komplikasi apa pun. Mereka hanya memiliki fungsi yang berfungsi.
Dan meskipun paket tersebut tidak memberikan detailnya kepada mereka, saya mencoba membuat format datanya cukup sederhana sehingga mereka dapat mengetahuinya jika saya tiba-tiba menghilang besok...
Kecepatannya meningkat secara nyata. Kami biasanya memindai fragmen genom yang signifikan secara fungsional. Sebelumnya, kami tidak dapat melakukan ini (ternyata terlalu mahal), namun sekarang, berkat struktur grup (bin) dan caching, permintaan untuk satu SNP rata-rata membutuhkan waktu kurang dari 0,1 detik, dan penggunaan data sangat besar. rendah sehingga biaya untuk S3 sangat murah.
Baru-baru ini saya mendapat perubahan dari 25+ TB data genotipe mentah untuk lab saya. Ketika saya mulai, menggunakan spark membutuhkan waktu 8 menit & biaya $20 untuk menanyakan SNP. Setelah menggunakan AWK+ untuk memprosesnya, sekarang dibutuhkan kurang dari sepersepuluh detik dan biayanya $10. Kepribadian saya menang.
— Nick Strayer (@NicholasStrayer)
Kesimpulan
Artikel ini sama sekali bukan panduan. Solusinya ternyata bersifat individual dan hampir pasti tidak optimal. Sebaliknya, ini adalah catatan perjalanan. Saya ingin orang lain memahami bahwa keputusan seperti itu tidak muncul sepenuhnya di kepala, melainkan hasil trial and error. Selain itu, jika Anda mencari data scientist, ingatlah bahwa menggunakan alat ini secara efektif memerlukan pengalaman, dan pengalaman memerlukan biaya. Saya senang bahwa saya mempunyai kemampuan untuk membayar, namun banyak orang lain yang dapat melakukan pekerjaan yang sama lebih baik dari saya tidak akan pernah mempunyai kesempatan karena kekurangan uang untuk mencoba.
Alat big data bersifat serbaguna. Jika Anda punya waktu, Anda hampir pasti dapat menulis solusi yang lebih cepat menggunakan teknik pembersihan, penyimpanan, dan ekstraksi data yang cerdas. Pada akhirnya, hal ini tergantung pada analisis biaya-manfaat.
Apa yang saya pelajari:
- tidak ada cara murah untuk mengurai 25 TB sekaligus;
- berhati-hatilah dengan ukuran file Parket Anda dan organisasinya;
- Partisi di Spark harus seimbang;
- Secara umum, jangan pernah mencoba membuat 2,5 juta partisi;
- Penyortiran masih sulit, begitu pula pengaturan Spark;
- terkadang data khusus memerlukan solusi khusus;
- Agregasi percikan cepat, tetapi partisi masih mahal;
- jangan tidur ketika mereka mengajari Anda dasar-dasarnya, seseorang mungkin sudah memecahkan masalah Anda di tahun 1980-an;
gnu parallel- ini adalah hal yang ajaib, setiap orang harus menggunakannya;- Spark menyukai data yang tidak terkompresi dan tidak suka menggabungkan partisi;
- Spark memiliki terlalu banyak overhead saat memecahkan masalah sederhana;
- Array asosiatif AWK sangat efisien;
- Anda dapat menghubungi
stdinиstdoutdari skrip R, dan karenanya menggunakannya dalam pipeline; - Berkat penerapan jalur cerdas, S3 dapat memproses banyak file;
- Alasan utama membuang-buang waktu adalah mengoptimalkan metode penyimpanan Anda sebelum waktunya;
- jangan mencoba mengoptimalkan tugas secara manual, biarkan komputer yang melakukannya;
- API harus sederhana demi kemudahan dan fleksibilitas penggunaan;
- Jika data Anda sudah dipersiapkan dengan baik, caching akan mudah!
Sumber: www.habr.com
