


Durante la pandemia attuale di COVID-19 sono emersi molti problemi su cui i hacker si sono lanciati con entusiasmo. Dai visori stampati in 3D e dalle mascherine fatte in casa fino alla sostituzione di un ventilatore polmonare meccanico completo, questo flusso di idee ha ispirato e deliziato. Allo stesso tempo, ci sono stati tentativi di progredire anche in un'altra area: la ricerca mirata a combattere direttamente il virus stesso.
A quanto pare, il più grande potenziale per fermare l'attuale pandemia e anticipare le successive risiede nell'approccio che cerca di affrontare la radice del problema. Questo approccio, di tipo «conosci il tuo nemico», è rappresentato dal progetto di calcolo Folding@Home. Milioni di persone si sono registrate al progetto e donano parte della potenza di calcolo dei loro processori e GPU, creando così il più grande [supercomputer distribuito] della storia.
Ma a cosa servono esattamente tutti questi exaflop? Perché è necessario investire tali potenze di calcolo nella ? Какая тут работает биохимия, зачем вообще белкам нужно укладываться? Вот краткий обзор фолдинга белков: что это, как он происходит и в чём его важность.
Per iniziare, la domanda più importante è: a cosa servono le proteine?
Le proteine sono strutture vitali. Non solo forniscono materiale da costruzione per le cellule, ma fungono anche da enzimi catalizzatori per praticamente tutte le reazioni biochimiche. Le proteine, che siano o , sono lunghe catene , disposti in una sequenza specifica. Le funzioni delle proteine sono determinate dalla posizione degli amminoacidi specifici all'interno della proteina. Per esempio, se una proteina deve legarsi a una molecola caricata positivamente, il sito di legame deve essere occupato da amminoacidi caricati negativamente.
Per comprendere come le proteine ottengano la struttura che determina la loro funzione, è necessario esaminare le basi della biologia molecolare e il flusso di informazioni nella cellula.
La produzione, o delle proteine inizia con il processo di . Durante la trascrizione, la doppia elica del DNA, che contiene le informazioni genetiche della cellula, si slega parzialmente, permettendo l'accesso alle basi azotate del DNA all'enzima chiamato . Il compito dell'RNA polimerasi è quello di realizzare una copia dell'RNA, o trascrizione, di un gene. Questa copia del gene, chiamata (mRNA), è una singola molecola perfettamente adatta a gestire le fabbriche proteiche all'interno della cellula, , che si occupano della produzione, o delle proteine.
I ribosomi funzionano come dispositivi di assemblaggio: catturano il modello di mRNA e lo abbinano ad altri piccoli frammenti di RNA, (tRNA). Ogni tRNA ha due siti attivi: una sezione di tre basi chiamata , che deve corrispondere ai codoni di mRNA appropriati, e una regione per il legame di un amminoacido specifico per questo . Durante la traduzione, le molecole di tRNA nel ribosoma cercano casualmente di legarsi all'mRNA tramite gli anticodoni. In caso di successo, la molecola di tRNA attacca il suo amminoacido a quello precedente, formando un ulteriore anello nella catena di amminoacidi codificata dall'mRNA.
Questa sequenza di amminoacidi rappresenta il primo livello della gerarchia strutturale di una proteina, e per questo motivo viene chiamata . L'intera struttura tridimensionale della proteina e le sue funzioni derivano direttamente dalla struttura primaria e dipendono dalle diverse proprietà di ciascun aminoacido e dalle loro interazioni reciproche. Senza queste proprietà chimiche e le interazioni tra aminoacidi, sarebbero rimasti sequenze lineari senza una struttura tridimensionale. Questo è evidente ogni volta che cuciniamo: durante questo processo avviene la della struttura tridimensionale delle proteine.
Legami a lungo raggio tra le parti delle proteine
Il livello successivo della struttura tridimensionale, che va oltre la primaria, è stato astutamente denominato . Essa comprende legami idrogeno tra aminoacidi di azione relativamente ravvicinata. La sostanza principale di queste interazioni stabilizzanti si riduce a due cose: e . L'alfa-elica forma una parte strettamente attorcigliata del polipeptide, mentre il beta-foglio rappresenta un'area liscia e ampia. Entrambe le strutture possiedono sia proprietà strutturali che funzionali, che dipendono dalle caratteristiche degli amminoacidi che le compongono. Per esempio, se l'alfa-elica è composta principalmente da amminoacidi idrofili, come o , è probabile che partecipi a reazioni acquose.
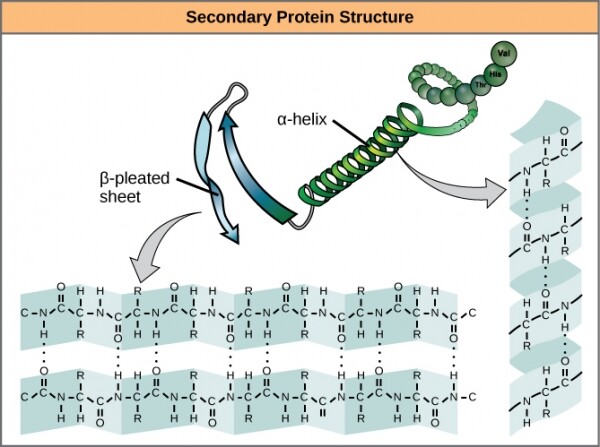
Alpha-eliche e beta-fogli nei proteine. I legami idrogeno si formano durante l'espressione delle proteine.
Queste due strutture e le loro combinazioni formano il successivo livello di struttura delle proteine — . A differenza dei semplici frammenti di struttura secondaria, la struttura terziaria è principalmente influenzata dall'idrofobicità. Nei centri della maggior parte delle proteine si trovano amminoacidi altamente idrofobici, come o , e l'acqua viene esclusa a causa della natura «grassa» dei radicali. Queste strutture spesso compaiono nelle proteine transmembrana, integrate nella doppia membrana lipidica che circonda le cellule. Le parti idrofobiche delle proteine rimangono termodinamicamente stabili all'interno della parte lipidica della membrana, mentre le sezioni idrofile della proteina sono esposte all'ambiente acquoso da entrambi i lati.
Inoltre, la stabilità delle strutture terziarie è garantita da interazioni a lungo raggio tra gli amminoacidi. Un esempio classico di tali legami è , che si verifica spesso tra due radicali di cisteina. Se, durante un trattamento di permanente in un salone di bellezza, hai percepito un odore che ricorda un po' le uova marce, allora si trattava di una parziale denaturazione della struttura terziaria della cheratina presente nei capelli, causata dalla riduzione dei legami disolfuro attraverso miscele miscele.
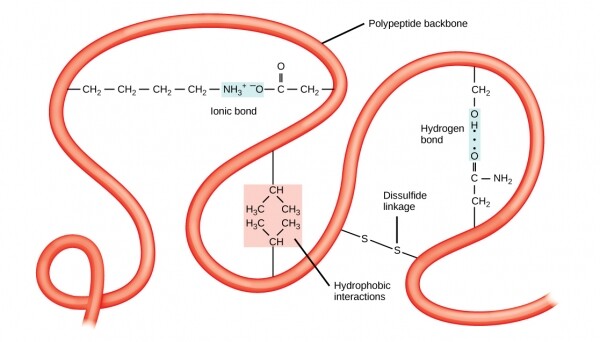
La struttura terziaria è stabilizzata da interazioni a lungo raggio, come idrofobicità o legami disolfuro.
I legami disolfuro possono formarsi tra radicali in una catena polipeptidica o tra cisteine di diverse catene complete. Le interazioni tra diverse catene formano livello della struttura proteica. Un ottimo esempio di struttura quaternaria è nel vostro sangue. Ogni molecola di emoglobina è composta da quattro globine identiche, parti di proteina, ognuna delle quali è mantenuta in una posizione specifica all'interno del polipeptide tramite ponti disolfuro, ed è anche legata a una molecola di eme contenente ferro. Tutte e quattro le globine sono legate insieme da ponti disolfuro inter-molecolari e l'intera molecola si lega contemporaneamente a più molecole di ossigeno, fino a quattro, e può rilasciarle all'occorrenza.
La modellazione delle strutture nella ricerca di trattamento per la malattia
Le catene polipeptidiche iniziano a ripiegarsi nella forma finale durante la traduzione, quando la catena in crescita esce dal ribosoma – simile a come un segmento di filo in lega a memoria può assumere forme complesse quando riscaldato. Tuttavia, come sempre in biologia, le cose non sono mai così semplici.
In molte cellule, prima della traduzione, i geni trascritti subiscono un editing significativo che altera notevolmente la struttura primaria della proteina rispetto alla sequenza di basi del gene originale. In questo processo, i meccanismi di traduzione spesso si avvalgono dell'aiuto di accompagnatori molecolari, proteine che si legano temporaneamente alla catena polipeptidica in via di sviluppo, impedendo così che assuma qualsiasi forma intermedia da cui non possano poi passare a quella finale.
Tutto ciò serve a sottolineare che prevedere la forma finale di una proteina non è un compito banale. Per decenni, l'unico modo per studiare la struttura delle proteine è stato attraverso metodi fisici come la cristallografia a raggi X. Solo alla fine degli anni '60, i biochimici fisici hanno iniziato a costruire modelli computazionali di ripiegamento delle proteine, concentrandosi principalmente sulla modellazione della struttura secondaria. Questi metodi e i loro successori richiedono enormi volumi di dati in ingresso oltre alla struttura primaria, come ad esempio tabelle degli angoli di legame degli amminoacidi, elenchi di idrofobicità, stati caricati e persino la conservazione della struttura e del funzionamento nel corso di periodi evolutivi — tutto ciò per cercare di indovinare come apparirà la proteina finale.
I metodi computazionali odierni per la previsione della struttura secondaria, come quelli utilizzati nella rete Folding@Home, raggiungono circa l'80% di precisione, un risultato piuttosto buono considerando la complessità della sfida. I dati ottenuti dai modelli predittivi su proteine come quella delle spine del SARS-CoV-2 saranno confrontati con i dati degli studi fisici sul virus. Alla fine, sarà possibile ottenere una struttura proteica precisa e, forse, capire come il virus si lega ai recettori. umano, presente nelle vie respiratorie che conducono all'interno del corpo. Se riusciremo a comprendere questa struttura, probabilmente troveremo farmaci in grado di bloccare il legame e prevenire l'infezione.
La ricerca sul folding delle proteine è al centro della nostra comprensione di un numero così elevato di malattie e infezioni che, anche quando con l'aiuto della rete Folding@Home riusciremo a trovare un modo per combattere il COVID-19, il cui aumento esplosivo stiamo osservando recentemente, questa rete non rimarrà inattiva a lungo. È uno strumento di ricerca eccellente per lo studio dei modelli proteici alla base di decine di malattie legate al folding errato delle proteine, come ad esempio la malattia di Alzheimer o una variante della malattia di Creutzfeldt-Jakob, spesso erroneamente chiamata 'mucca pazza'. E quando inevitabilmente apparirà un altro virus, saremo già pronti a combatterlo di nuovo.
Fonte: habr.com
