


Si të lexoni këtë artikull: kërkoj ndjesë që teksti doli kaq i gjatë dhe i çrregullt. Për të kursyer kohën tuaj, çdo kapitull e filloj me një hyrje 'Çfarë kam mësuar', ku përmbledh në një ose dy fjalitë thelbin e kapitullit.
‘Thjesht tregoni zgjidhjen!’ Nëse thjesht dëshironi të shihni se çfarë arrita, kaloni në kapitullin 'Po bëhem më shpikës', por mendoj se është më interesante dhe e dobishme të lexoni për dështimet.
Së fundmi, më është dhënë detyra të themeloj një proces për përpunimin e një sasie të madhe të sekuencave të ADN-së (teknikisht, ky është një SNP-chip). Duhej të merrja shpejt të dhëna mbi një vendndodhje gjenetike të caktuar (e njohur si SNP) për modelimin e mëvonshëm dhe detyra të tjera. Me ndihmën e R dhe AWK, arrita të pastroj dhe organizoj të dhënat në një mënyrë natyrale, duke acceleruar dukshëm përpunimin e kërkesave. Nuk ishte e lehtë dhe kërkoi shumë iteracione. Ky artikull do t'ju ndihmojë të shmangni disa nga gabimet e mia dhe do të tregojë se çfarë arrita përfundimisht.
Për të filluar, disa shpjegime hyrëse.
Të dhënat
Qendra jonë universitare për përpunimin e informacionit gjenetik na ofroi të dhënat në një format TSV me një kapacitet prej 25 TB. Më janë dorëzuar të ndara në 5 paketa, të kompresuara me Gzip, secili prej të cilëve përmbante rreth 240 skedarë katër gigabajtësh. Çdo rresht kishte të dhëna për një SNP të një personi. Në total, u transmetuan të dhëna për rreth 2.5 milion SNP dhe rreth 60 mijë persona. Përveç informacionit mbi SNP në skedarë, kishte shumë kolona me numra që reflektonin karakteristika të ndryshme, si intensiteti i leximit, frekuenca e aleleve të ndryshme, etj. Në total, kishte rreth 30 kolona me vlera unike.
Qëllimi
Si në çdo projekt të menaxhimit të të dhënave, më e rëndësishmja ishte të përcaktohej se si do të përdoren të dhënat. Në këtë rast ne kryesisht do të përshtatim modelet dhe proceset e punës për SNP-në bazuar në SNP. Kështu, në të njëjtën kohë, ne do të na nevojiten të dhëna vetëm për një SNP. Duhej të mësoja si të nxjerr sa më lehtë, shpejt dhe me kosto më të ulët të gjitha të dhënat që i përkasin një nga 2.5 milion SNP.
Si të mos e bëni këtë
Do ta citoj një klişe të përshtatshme:
Unë nuk kam dështuar njëmijë herë, thjesht kam zbuluar njëmijë mënyra për të mos nxjerrë një sasi të madhe të dhënash në një format të përshtatshëm për kërkesa.
Prova e parë
Çfarë kam mësuar: nuk ka një mënyrë të lirë për të nxjerrë 25 TB njëherësh.
Pas dëgjuar lëndën "Metodat Avancuara të Përpunimit të të Dhënave të Mëdha" në Universitetin Vanderbilt, isha i sigurt se ishte e thjeshtë. Ndoshta do të duheshin një orë ose dy për të konfiguruar një server Hive për të kaluar të gjitha të dhënat dhe për të raportuar rezultatin. Sepse të dhënat tona ruhen në AWS S3, përdora shërbimin , i cili lejon të aplikosh kërkesa Hive SQL për të dhënat S3. Nuk është e nevojshme të konfigurosh/ejndështosh një klaster Hive, dhe paguan vetëm për të dhënat që kërkon.
Pasi i tregova Athënës të dhënat e mia dhe formatin e tyre, ekzekutova disa teste me kërkesa të ngjashme:
select * from intensityData limit 10;Dhe shpejt mora rezultate të strukturuara mirë. E gatshme.
Derisa nuk e provuam të përdornim të dhënat në punë…
Më kërkuan të nxjerr të gjitha informacionet për SNP, për ta testuar modelin mbi të. E lançova kërkesën:
select * from intensityData
where snp = 'rs123456';…dhe fillova të prisja. Pasi kaluan tetë minuta dhe më shumë se 4 TB të dhënash të kërkuara, mora rezultatin. Athena ngarkon për volumet e të dhënave të gjetura, me $5 për terabajt. Pra, kjo kërkesë e vetme më kushtoi $20 dhe tetë minuta pritje. Për të përpunuar modelin në të gjitha të dhënat, do të duhet të prisja 38 vjet dhe të paguaj $50 milion. Është e qartë se këtë ne nuk mund ta përballonim.
Duhej të përdorja Parquet…
Çfarë kam mësuar: kini kujdes me madhësinë e skedarëve tuaj Parquet dhe organizimin e tyre.
Fillimisht përpiqesha të rregulloja situatën, duke konvertuar të gjitha TSV në . Ato janë të përshtatshme për punë me grupe të mëdha të dhënash, sepse informacioni në to ruhet në formë kolonash: çdo kolonë gjendet në segmentin e vet të memories/diskut, përkundrazi me skedarët tekstorë, në të cilët rreshtat përmbajnë elementet e çdo kolone. Dhe nëse duhet të kërkoni diçka, mjafton të lexoni kolonën përkatëse. Për më tepër, në çdo skedar në kolonë ruhet një gamë e vlerave, kështu që nëse vlera e kërkuar nuk ndodhet brenda gamës së kolonës, Spark nuk do të humbasë kohë duke skanuar të gjithë skedarin.
Kisha nisur një detyrë të thjeshtë për të konvertuar TSV-të tona në Parquet dhe ngarkoha skedarë të rinj në Athena. Kjo zuri rreth 5 orë. Por kur e nisa pyetjen, ekzekutimi i saj mori përafërsisht të njëjtën kohë dhe pak më pak para. Arsyetimi është se Spark, duke përpjekur të optimizojë detyrën, thjesht shpaktoi një TSV-çank dhe e vendosi atë në çankun e tij Parquet. Dhe për shkak se çdo çank ishte mjaft i madh dhe përmbante të dhëna të plota të shumë njerëzve, çdo skedar përmbante të gjitha SNP-të, kështu që Spark duhej të hapej të gjithë skedarët për të nxjerrë informacionin e nevojshëm.
Është interesante se tipi i kompresimit që përdoret si default (dhe i rekomanduar) në Parquet — snappy, — nuk është i ndarë (splitable). Prandaj, çdo ekzekutor (executor) ngeci në detyrën e shpaketimit dhe ngarkimit të setit të dhënash të plotë në 3.5 GB.
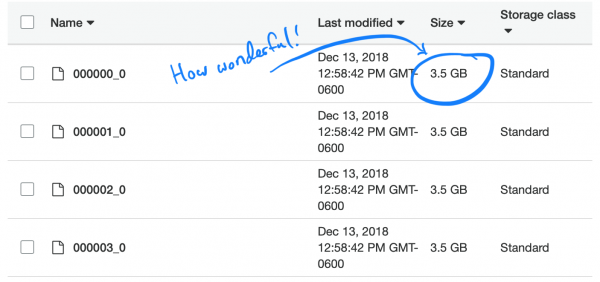
Po shqyrtojmë problemin
Çfarë kam mësuar: është e vështirë të renditësh, veçanërisht kur të dhënat janë të shpërndara.
Më dukej se tani e kuptova thelbin e problemit. Më duhej vetëm të rendisja të dhënat sipas kolonës SNP, e jo sipas njerëzve. Atëherë në një copë të dhënash do të ruhen disa SNP, dhe atëherë funksioni "i zgjuar" i Parquet do të shfaqet në të gjithë shkëlqimin e tij "hap vetëm nëse vlera është brenda gamës".
Unë duke ndjekur klasën e algoritmeve në universitet: «Ugh, askush nuk i intereson kompleksiteti kompjutor i të gjitha këtyre algoritmeve të renditjes»
Unë duke u përpjekur të rendis në një kolonë në një 20TB tabelë: «Pse po zgjat kaq shumë?» vështirësi.
— Nick Strayer (@NicholasStrayer)
AWS me siguri nuk do të donte t'i kthejë paratë për shkak të «Unë jam një student i përhumbur». Pas lançimit të renditjes në Amazon Glue, ajo punoi për 2 ditë dhe përfundoi me dështim.
Çfarë është për partitë?
Çfarë kam mësuar: partitë në Spark duhet të jenë të balancuara.
Pastaj më erdhi ideja për të ndarë të dhënat në kromozome. Janë 23 copë (dhe disa të tjera, nëse llogarisim ADN-në mitokondriale dhe zonat e paqarta (unmapped)).
Kjo do të lejojë ndarjen e të dhënave në porcione më të vogla. Nëse e shtojmë në funksionin eksportues të Spark në skenarin Glue vetëm një rresht partition_by = "chr", atëherë të dhënat duhet të ndahen në buxhetet (buckets).
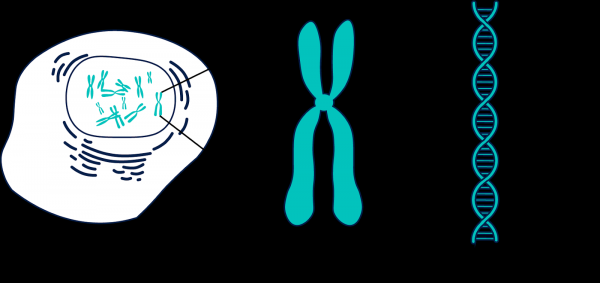
Gjeni është përbërë nga fragmente të shumta, të quajtura kromozome.
Fatkeqësisht, kjo nuk funksionoi. Kromozomet kanë madhësi të ndryshme, që do të thotë edhe sasi të ndryshme informacioni. Kjo do të thotë që detyrat që Spark dërgonte tek punëtorët nuk ishin të balancuara dhe u kryen ngadalë, sepse disa nyje përfundonin më herët dhe qëndronin të papuna. Megjithatë, detyrat u realizuan. Por kur kërkohej një SNP, mosbalancimi përsëri u bë shkak për probleme. Kostoja e përpunimit të SNP në kromozomet më të mëdha (dmth atje ku ne duam të marrim të dhënat) u zvogëlua vetëm rreth 10 herë. Shumë, por jo mjaftueshëm.
Dhe nëse e ndajmë në pjesë edhe më të vogla?
Çfarë kam mësuar: asnjëherë mos u përpoqni të bëni 2.5 milion pjesë.
Unë vendosa të shkoj deri në fund dhe bëra ndarjen për çdo SNP. Kjo garantoi madhësi të njëjtë për pjesët. ISHTE NJË IDE E KEQE. Unë përdora Glue dhe shtova një rresht të pafajshëm partition_by = 'snp'. Detyra filloi dhe nisi të ekzekutohet. Një ditë më vonë kontrollova dhe pashë se në S3 ende nuk kishte asgjë të regjistruar, kështu që e ndalova detyrën. Duket se Glue po regjistronte skedarë të përkohshëm në një vend të fshehtë në S3, dhe shumë skedarë, ndoshta disa miliona. Si pasojë, gabimi im më kushtoi më shumë se një mijë dollarë dhe nuk e gëzoi mentorin tim.
Kategoritë + renditja
Çfarë kam mësuar: renditja ende është e vështirë, ashtu si dhe konfigurimi i Spark.
Akhia e fundit e kategorive ishte ajo që unë kategoritizova kromozomet dhe pastaj rendita secilën kategori. Në teori, kjo do të lejonte të përshpejtonte çdo kërkesë, sepse të dhënat e dëshiruara mbi SNP duhet të ishin brenda disa blloqeve Parquet në një gamë të caktuar. Fatkeqësisht, renditja e të dhënave të kategorizuara ishte një detyrë e vështirë. Si rezultat, kalova në EMR për një grup të personalizuar dhe përdora tetë instanca të fuqishme (C5.4xl) dhe Sparklyr për të krijuar një proces pune më fleksibël…
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)…megjithatë, detyra nuk u krye. Kam provuar në mënyra të ndryshme: kam rritur shpërndarjen e memories për secilin ekzekutor të kërkesave, kam përdorur nodo me memorie më të madhe, kam aplikuar variabla të transmetimit (broadcasting variables), por çdo herë rezultoi se ishin masa të përkohshme, dhe gradualisht ekzekutorët filluan të dështonin, deri sa gjithçka u ndal.
Përditësim: kështu fillon.
— Nick Strayer (@NicholasStrayer)
Po bëhem më shpikës
Çfarë kam mësuar: ndonjëherë të dhënat e veçanta kërkojnë zgjidhje të veçanta.
Çdo SNP ka një vlerë pozite. Ky numër i korrespondon sasisë së bazave që ndodhen përgjatë kromozomit të tij. Kjo është një mënyrë e mirë dhe natyrale për të organizuar të dhënat tona. Në fillim doja të partitionoja sipas rajoneve të çdo kromozomi. Për shembull, pozitat 1 - 2000, 2001 - 4000 etj. Por problemi është se SNP-të janë të shpërndara në kromozome në mënyrë të pabarabartë, kështu që madhësia e grupeve do të jetë shumë e ndryshme.
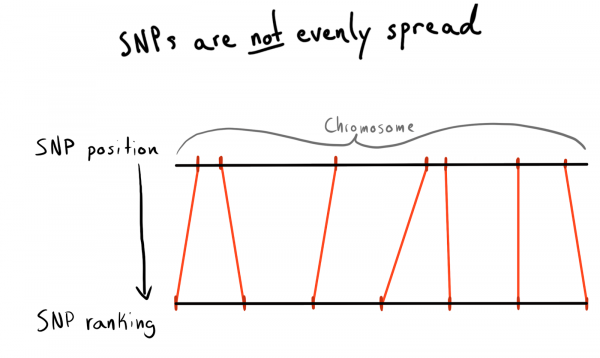
Si pasojë, arrita në ndarjen e pozita në kategori (rank). Duke përdorur të dhënat e ngarkuara tashmë, e kalova një kërkesë për të marrë listën e SNP-ve unike, pozitat e tyre dhe kromozomet. Më pas, i rendita të dhënat brenda çdo kromozomi dhe grupova SNP-të në grupe (bin) me madhësi të caktuar. Le ta themi, 1000 SNP për grup. Kjo më dha lidhjen e SNP-ve me grupin në kromozom.
Në fund, kreva grupet (bin) me 75 SNP, arsyeja do të shpjegohet më poshtë.
snp_to_bin %
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()Përpjekja e parë me Spark
Çfarë kam mësuar: bashkimi në Spark funksionon shpejt, por ndarja ende kushton shumë.
Donim të lexonim këtë kuadër të vogël (2.5 milion rreshta) në Spark, ta bashkonim atë me të dhënat e papërpunuara dhe pastaj ta ndarim sipas kolonës së sapo shtuar. bin.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
) Unë kam përdorur sdf_broadcast(), kështu që Spark mëson se duhet ta dërgojë kuadrin e të dhënave në të gjitha nyjat. Kjo është e dobishme nëse të dhënat janë të vogla dhe kërkohen për të gjitha detyrat. Ndryshe, Spark përpiqet të jetë inteligjent dhe shpërndan të dhënat sipas nevojës, çka mund të shkaktojë ngadalësime.
Dhe me sërish ideja ime nuk funksionoi: detyrat punonin për një kohë, përfundonin bashkimin dhe pastaj, siç ndodhte me ekzekutorët e nisur nga ndarje, filluan të dështojnë.
Po shtoj AWK
Çfarë kam mësuar: mos flini kur ju mësojnë bazat. Me siguri dikush e ka zgjidhur problemin tuaj që në vitet 1980.
Derisa arsyetimi i dështimeve të mia me Spark deri në këtë moment ishte çrregullimi i të dhënave në klasër, ndoshta situata mund të përmirësohet me përpunimin paraprak. Vendosa të provoj të ndaja të dhënat e papërpunuara tekstuale në kolona kromozomesh, me shpresën se do t'i ofroja Spark’ut të dhëna "të paraparë për ndarje".
Kërkova në StackOverflow se si të ndahet sipas vlerave të kolonave dhe gjeta Me ndihmën e AWK mund të ndash një skedar teksti sipas vlerave të kolonave, duke shkruar një skenar dhe jo duke dërguar rezultatet në stdout.
Për provë, shkrova një skenar Bash. Shkarkova një nga skedat TSV të paketuara, pastaj e shpërndava me gzip dhe e dërgova në awk.
gzip -dc path/to/chunk/file.gz |
awk -F 't'
'{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'Kjo funksionoi!
Plotësimi i bërthamave
Çfarë kam mësuar: gnu parallel — është një gjë magjike, të gjithë duhet ta përdorin.
Ndërrimi po kalonte mjaft ngadalë, dhe kur hodha në punë htop, për të verifikuar përdorimin e një instancë të fuqishme (dhe të shtrenjtë) EC2, u zbulua se isha duke përdorur vetëm një bërthamë dhe rreth 200 MB memorie. Për të zgjidhur problemin dhe për të mos humbur shumë para, duhej të mendoja se si ta ndaja punën. Fatmirësisht, në një libër të mrekullueshëm të Jeron Janssens, gjetja e një kapitulli që i dedikohej ndarjes së punës. Prej tij mësova për gnu parallel, një metodë shumë fleksibël për implementimin e shumë-threading në Unix.

Kur hodha në punë ndarjen me procesin e ri, gjithçka ishte mirë, por kishte një ngushticë — shkarkimi i objekteve S3 në disk nuk ishte shumë i shpejtë dhe nuk ishte plotësisht i ndarë. Për ta rregulluar këtë, bëra këtë:
- Zgjodha se mund të implementoja fazën e shkarkimit S3 pikërisht në pipeline, duke eliminuar plotësisht ruajtjen ndërmjet disk. Kjo do të thotë se mund të shmang hashurrejten e të dhënave të papërpunuara në disk dhe të përdor një ruajtje edhe më të vogël, për rrjedhojë edhe më të lirë në AWS.
- Me komandën
aws configure set default.s3.max_concurrent_requests 50kaq shumë rriti numrin e rrjedhave që përdor AWS CLI (normalisht janë 10). - Kalova në një instancë EC2 të optimizuar për shpejtësi rrjeti, me shkronjën n në emër. Pashë që humbja e fuqisë kompjuterike kur përdorja instancat n kompensohej me bollëk nga rritja e shpejtësisë së ngarkesës. Për shumicën e detyrave përdora c5n.4xl.
- Ndryshova
gzipnë , është një mjet gzip, i cili di të bëjë gjëra të mrekullueshme për të bërë paralelizimin e detyrave që fillimisht nuk janë paralelizuar, siç është dekompresimi i skedarëve (kjo ndihmoi më pak se çdo gjë tjetër).
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe
"awk -F 't' '{print $1",..."$30">"chunked/{#}_chr"$15".csv"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneKëto hapa janë kombinuar për të punuar shumë shpejt. Falë rritjes së shpejtësisë së shkarkimit dhe heqjes së shkrimit në disk, tani mund të përpunoj një paketë prej 5 terabajtësh për vetëm disa orë.
Nuk ka gjë më të bukur se të shohësh të gjithë bërthamat që po paguan për AWS duke u përdorur. Falë gnu-parallel mund të dekompresoj dhe ndaj një csv 19gig po aq shpejt sa e shkarkoj. Nuk mund të bëja as që spark të funksiononte.
— Nick Strayer (@NicholasStrayer)
Ky tweet duhet të kishte përmendur 'TSV'. Më vjen keq.
Përdorimi i të dhënave të riparparuara
Çfarë kam mësuar: Spark e do të dhënat e papaketuara dhe nuk i pëlqen të kombinojë partitë.
Tani tani tani janë ruajtur në S3 në një format të papaketuara (lexo, të ndara) dhe gjysmë të renditur, dhe unë mund të kthehem përsëri te Spark. Më priste një surprizë: prapë nuk arrita të arrija dëshirën! Ishte shumë e vështirë të thosha saktë se si ishin paritizuar të dhënat. Edhe kur e bëra këtë, rezultoi se kishte shumë paritë (95 mijë), dhe kur unë me anë të coalesce e gjithësova numrin e tyre në kufij të arsyeshëm, kjo prishi paritizimin tim. Jam i sigurt se kjo mund të korrigjohet, por pas disa ditëve kërkimesh nuk arrita të gjej zgjidhje. Në fund të fundit, përfundova të gjitha detyrat në Spark, megjithëse këtë e mori pak kohë, dhe skedarët e mi Parquet ishin jo shumë të vegjël (~200 Kb). Megjithatë, të dhënat ishin aty ku duheshin.
Shumë të vogla dhe të ndryshme, mrekullueshëm!
Testimi i pyetjeve lokale në Spark
Çfarë kam mësuar: në Spark ka shumë shpenzime kur zgjidhni detyra të thjeshta.
Duke të dhënat në një format të menduar, arrita të testoj sh brin. Konfigurova skriptin në R për të nisur një server lokal Spark dhe më pas ngarkova një kornizë të dhënash Spark nga depoja e caktuar të grupeve Parquet (bin). Përpiqesha të ngarkoja të gjitha të dhënat, por nuk arrita ta bëj Sparklyr të njohë ndarjen.
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Filloni një kronometër
start_time <- Sys.time()
# Ngarko binin e dëshiruar në Spark
intensity_data %
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Ndarja e binit në snp dhe pastaj mbledhja në lokal
test_subset %
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)Ekzekutimi mori 29,415 sekonda. Shumë më mirë, por jo aq mirë për testimin masiv të ndonjë gjëje. Për më tepër, nuk mund të përshpejtoja funksionimin me ndihmën e caching, sepse kur përpiqesha të cache në kujtesë kornizën e të dhënave, Spark gjithmonë binte, madje edhe kur i ndava më shumë se 50 GB memorie për datasetin që kishte peshë më pak se 15.
Kthimi te AWK
Çfarë kam mësuar: array të asociuar në AWK janë shumë efikase.
E dija se mund të arrija një shpejtësi më të madhe. Më kujtohej se në udhëzimin e shkëlqyer Lexova për një karakteristikë të shkëlqyer, e cila quhet "". Në thelb, këto janë çiftet çelës-vlerë, të cilat për një arsye të caktuar në AWK janë quajtur ndryshe, dhe prandaj nuk e kam kujtuar shumë. më kujtoi se termi "marrëveshje asocative" është shumë më i vjetër se termi "çift çelës-vlerë". Edhe nëse ju , nuk do ta gjeni atë, megjithatë do të gjeni marrëveshje asocative! Për më tepër, "çifti çelës-vlerë" shpesh lidhet me bazat e të dhënave, prandaj është shumë më logjike të krahasohet me hashmap. Kuptova se mund të përdor këto marrëveshje asocative për të lidhur SNP-të e mia me tabelën e grupeve (bin table) dhe të dhënat e papërpunuara pa përdorur Spark.
Për këtë në skriptin AWK përdora bllokun FILLIM. Ky është një fragment kodi që ekzekutohet para se rreshti i parë i të dhënave të kalojë në trupin kryesor të skriptit.
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
} Ekipa while(getline...) ngarkohet të gjitha rreshtat nga CSV i grupit (bin), duke caktuar kolonën e parë (emri SNP) si çelës për marrëveshjen asocative bin dhe vlera e dytë (grupi) si një vlerë. Pastaj në bllokun { }, i cili ekzekutohet për të gjitha rreshtat e skedarit kryesor, secili rresht dërgohet në skedarin e daljes, që merr një emër unik në varësi të grupit të tij (bin): ..._bin_"bin[$1]"_....
Variablat numri_batch dhe id_blloku përputhen me të dhënat e ofruara nga tubacioni, duke shmangur kështu gjendjen e garës, dhe çdo rrjedhë ekzekutimi e nisur paralel, shkruante në skedarin e tij unik.
Pasi të gjitha të dhënat e dështuara i kam shpërndarë në dosje sipas kromozomëve, që mbeteshin pas eksperimentit tim të mëparshëm me AWK, tani mund të shkruaj një skenar tjetër Bash, për të përpunuar një kromozom njëherësh dhe të dorëzoj në S3 të dhëna më të ndara.
DESIRED_CHR='13'
# Shkarko të dhënat e kromozomeve nga s3 dhe ndaj ato në bins
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'duke lexuar {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=""$DESIRED_CHR"" -v chunk="{}" -f split_on_chr_bin.awk"
# Kombino të gjitha blloqet e proceseve paralele në skedarë të vetme dhe ngarko në rds duke përdorur R
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'duke zipuar bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/* Në skenar ka dy seksione paralel.
Në seksionin e parë, lexohen të dhënat nga të gjitha skedarët që përmbajnë informacion për kromozomin e nevojshëm, pastaj këto të dhëna shpërndahen në kanale, të cilat ndihmojnë në organizimin e skedarëve në grupe përkatëse (bin). Për të shmangur ndodhitë e garave, kur disa kanale shkruajnë në të njëjtin skedar, AWK i dërgon emrat e skedarëve për të shkruar të dhënat në vende të ndryshme, për shembull, chr_10_bin_52_batch_2_aa.csv. Si rezultat, krijohet një mori skedarësh të vegjël në disk (për këtë përdora volume EBS një terabajtësh).
Pauza e seksionit të dytë paralel kalon përmes grupeve (bin) dhe bashkon skedarët e tyre të veçantë në CSV të përbashkët me cat, dhe pastaj i dërgon ata për eksport.
Translimi në R?
Çfarë kam mësuar: është e mundur të aksesoni stdin dhe stdout nga skripti R, dhe për këtë arsye ta përdorni atë në pauzë.
Në skriptin Bash, mund të keni vërejtur këtë rresht: ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... Ai translem any skedarë të bashkuar të grupit (bin) në skriptin R më poshtë. {} është një metodologji speciale paralel, e cila çdo të dhënë që e dërgon në kanalin përkatës e fut drejtpërdrejt në komandën vetë. Opsioni {#} siguron një ID unik të ekzekutimit në kanal, dhe {%} përfaqëson numrin e slotit të detyrës (në përsëritje, por kurrë njëkohësisht). Lista e të gjitha opsioneve mund të gjendet në
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
) Kur variabli file("stdin") dërgohet në readr::read_csv, të dhënat, të transkriptuara në skenarin R, ngarkohen në një kuadër që më pas në formën e .rds-skedarit me ndihmën e aws.s3 shkruhet drejtpërdrejt në S3.
RDS është diçka si një version i vogël i Parquet, pa rafinamentet e depove kolonike.
Pas përfundimit të skenarit Bash, kam marrë një grup .rds-skedarësh të vendosur në S3, që më dha mundësinë të përdor kompresim efektiv dhe lloje të integruara.
Pavarësisht përdorimit të R të ngadalshëm, gjithçka funksionoi shumë shpejt. Nuk është për t'u habitur që fragmentet në R që merren me leximin dhe shkrimin e të dhënave janë optimizuar mirë. Pas testimit në një kromozom me madhësi mesatare, detyra u krye në instancën C5n.4xl për rreth dy orë.
Kufizimet e S3
Çfarë kam mësuar: përmes implementimit të zgjuar të rrugëve, S3 mund të обработojë shumë skedarë.
Isha i shqetësuar nëse S3 do të mund të përballonte shumë skedarë që i ishin kaluar. Mund të bëja emrat e skedarëve me kuptim, por si do të kërkonte S3 për ta?

Kërkesat në S3 janë thjesht për estetikë, në të vërtetë sistemi nuk është i interesuar për simbole. /.
Duket se S3 paraqet rrugën për një skedar të caktuar si një çelës të thjeshtë në një tabelë hesh apo një bazë të dhënash dokumentesh. Një bucket mund të konsiderohet si një tabelë, ndërsa skedarët - si regjistra në këtë tabelë.
Duke qenë se shpejtësia dhe efikasiteti janë të rëndësishme për fitimin në Amazon, nuk është befasi që ky sistem "çelës-në-vlerën-e-rrugës-për-skedarin" është jashtëzakonisht i optimizuar. Kam përpjekur të gjej një ekuilibër: që të mos nevojitet të bëhen shumë kërkesa get, por që kërkesat të ekzekutohen shpejt. Doli se më mirë është të krijohen rreth 20,000 skedarë binarë. Mendoj se nëse vazhdoj të optimizoj, mund të arrij një rritje të shpejtësisë (për shembull, duke krijuar një bucket special për të dhënat, kështu duke ulur madhësinë e tabelës së kërkimit). Por nuk kishte më kohë dhe para për eksperimente të tjera.
Çfarë ndodh me përputhshmërinë e kryqëzuar?
Çfarë mësova: arsyeja kryesore për humbjen e kohës është optimizimi i parakohshëm i metodës suaj të ruajtjes.
Në këtë moment, është shumë e rëndësishme të pyesni veten: "Pse të përdorim një format dosje proprietar?" Arsyet qëndrojnë në shpejtësinë e ngarkesës (skedarët gzip CSV u ngarkuan 7 herë më ngadalë) dhe shkallën e përputhshmërisë me proceset tona të punës. Mund të rishikoj vendimin tim nëse R do të jetë në gjendje të ngarkojë lehtësisht skedarët Parquet (ose Arrow) pa barrë nga Spark. Në laboratorin tonë, të gjithë përdorin R, dhe nëse do të kem nevojë për të transformuar të dhënat në një format tjetër, unë ende kam të dhënat origjinale tekstuale, kështu që thjesht mund të rifilloj procesin.
Ndarja e punës
Çfarë kam mësuar: mos përpiquni të optimizoni detyrat manualisht, lëreni këtë t'i bëjë kompjuteri.
Kam rregulluar procesin e punës për një kromozom, tani duhet të përpunoj të dhënat e tjera.
Dëshiroja të ngrija disa instanca EC2 për transformim, por në të njëjtën kohë kam frikë se do të merrja një ngarkesë shumë të paekuilibruar në detyra të ndryshme për përpunim (ashtu si Spark vuante nga parti të paekuilibruara). Për më tepër, nuk isha i kënaqur të ngrija një instancë për çdo kromozom, pasi për llogaritë AWS ka një kufizim të paracaktuar prej 10 instancash.
Atëherë vendosa të shkruaj një skript R për optimizimin e detyrave të përpunimit.
Fillimisht kërkova që S3 të llogaritë sa hapësirë në ruajtje zë çdo kromozom.
library(aws.s3)
library(tidyverse)
chr_sizes %
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Nxjerr kromozomin nga emri i skedës
chr = str_extract(Key, 'chr.{1,4}.csv') %>%
str_remove_all('chr|.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Pjesëto për t'u marrë vlera në GB
# Një tibble: 27 x 2
chr total_size
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … me 17 rreshta më shumë Më pas shkrova një funksion që merr madhësinë totale, përzier rendin e kromozomeve, i ndan ato në grupe num_jobs dhe raporton se sa ndryshojnë madhësitë e të gjitha detyrave përpunuese.
num_jobs <- 7
# Sa e madhe do të ishte secila punë nëse do të ndahej perfekt?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job %
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)
# Një tibble: 1 x 2
sd data
1 153.Më pas, e kalova një mijë përzierje me purrr dhe zgjodha më të mirën.
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1) Kështu e mora një grup detyrash që ishin shumë të ngjashme në madhësi. Pastaj mbetej vetëm të mbështillja skriptin tim të mëparshëm të Bash në një cikël të madh. për. Shkruajtja e kësaj optimizimi mori rreth 10 minuta. Dhe kjo është shumë më pak se sa do të kisha shpenzuar për të krijuar detyrat manualisht në rast të pabarazisë së tyre. Prandaj mendoj se me këtë optimizim paraprak nuk gabova.
për DESIRED_CHR në "16" "9" "7" "21" "MT"
bëj
# Kodi për përpunimin e një kromozomi të vetëm
fiNë fund shtoj komandën e fikjes:
sudo shutdown -h now … dhe gjithçka funksionoi! Me ndihmën e AWS CLI ngrita instance dhe përmes opsionit user_data u jepja atyre skriptet e Bash të detyrave për përpunim. Ato u ekzekutuan dhe u fikën automatikisht, kështu që nuk paguajta për fuqi përpunuese të tepërt.
aws ec2 run-instances ...
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]"
--user-data file://<<job_script_loc>>Le të ambalazhojmë!
Çfarë kam mësuar: API duhet të jetë i thjeshtë për thjeshtësinë dhe fleksibilitetin e përdorimit.
Finalmente mora të marr të dhënat në vendin dhe formën e duhur. Duhej ta thjeshtoja maksimalisht procesin e përdorimit të të dhënave, në mënyrë që kolegët e mi të kishin më të lehtë. Dëshiroja të krijoja një API të thjeshtë për krijimin e kërkesave. Nëse në të ardhmen vendos të kaloj në .rds në skedarët Parquet, atëherë kjo duhet të jetë një çështje për mua dhe jo për kolegët. Për këtë, vendosa të krijoja një paketë të brendshme R.
Këtu mbledha dhe dokumentova një paketë të thjeshtë, që përmban vetëm disa funksione për qasje në të dhëna, të mbledhura rreth funksionit get_snp. Gjithashtu krijova një faqe për kolegët , në mënyrë që ata të mund të shohin lehtësisht shembuj dhe dokumentacion.
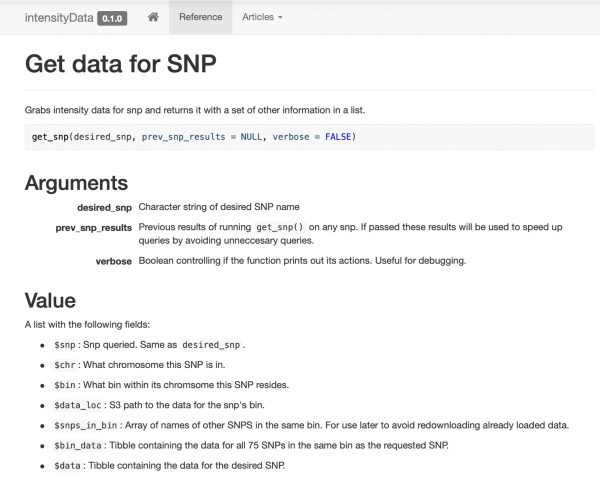
Keshilimi inteligjent
Çfarë kam mësuar: nëse të dhënat tuaja janë përgatitur mirë, do të jetë e lehtë të keshiloni!
Pasi një nga proceset kryesore aplikonte të njëjtin model analize në paketën SNP, vendosa të përdor grupimin në interesin tim. Kur dërgohet të dhëna për SNP, informacioni nga grupi gjithashtu bashkëngjitet objektit të kthyer (bin). Kjo do të thotë se kërkesat e vjetra mund (në teori) të shpejtojnë procesin e kërkesave të reja.
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
... Gjatë ndërtimit të paketës, kam ekzekutuar shumë benchmark-e për të krahasuar shpejtësinë duke përdorur metoda të ndryshme. Rekomandoj të mos neglizhohet këtë, sepse ndonjëherë rezultatet mund të jenë të papritura. Për shembull, dplyr::filter u tregua shumë më i shpejtë se kapja e rreshtave përmes filtrimit me bazë indeksimi, ndërsa marrja e një kolone nga kadri i të dhënave të filtruar punonte shumë më shpejt se aplikimi i sintaksës së indeksimit.
Vini re se objekti prev_snp_results përmban çelësin snps_in_bin. Ky është një array i të gjithë SNP-ve unikë në grup (bin), që lejon një kontroll të shpejtë për të parë nëse tashmë ka të dhëna nga kërkesa e mëparshme. Gjithashtu, e bën më të lehtë kalimin ciklik në të gjitha SNP-të në grup (bin) me këtë kod:
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}Rezultatet
Tani mund të (dhe kemi filluar seriozisht) të ekzekutojmë modele dhe skenare, të cilat më parë nuk ishin të aksesueshme. E mira më e madhe është se kolegët e mi në laborator nuk kanë nevojë të mendojnë për ndonjë kompleksitet. Ata thjesht kanë një funksion që punon.
Dhe ndërsa paketa i çliron ata nga detajet, unë përpiqesha të bëja formatin e të dhënave mjaft të thjeshtë për t'u kuptuar, nëse nesër ndonjëherë do të zhdukesha...
Shpejtësia është rritur dukshëm. Zakonisht ne skanojmë fraksione funksionalisht të rëndësishme të genomit. Më parë nuk mund ta bënim këtë (në mënyrë që ishte shumë e shtrenjtë), por tani, falë strukturës grupore (bin) dhe keshimit, për çdo kërkesë të një SNP shpenzohet mesatarisht më pak se 0.1 sekonda, dhe përdorimi i të dhënave është aq i ulët sa që shpenzimet për S3 janë minimale.
Së fundmi, më është ngarkuar menaxhimi i mbi 25 TB të dhënash të pa sistemizuar për gjenotipizimin në laboratorin tim. Kur fillova, përdorimi i spark merrte 8 minuta dhe kushtonte 20 dollarë për të kërkuar një SNP. Pas përdorimit të AWK + për procesim, tani merr më pak se një të dhjetën e një sekonde dhe kushton 0.00001 dollarë. Fitimi im personal i madh.
— Nick Strayer (@NicholasStrayer)
Përfundimi
Kyçja e këtij artikulli nuk është një udhëzues. Zgjidhja ka rezultuar e individualizuar dhe pothuajse me siguri jo optimale. Më mirë do ishte ta shihnim si një rrëfim të një udhëtimi. Dëshiroj që të tjerët të kuptojnë se zgjidhje të tilla nuk shfaqen krejtësisht të formuara në mendje, ato janë rezultat i provave dhe gabimeve. Përveç kësaj, nëse po kërkoni një specialist në analiza të dhënash, mbani parasysh se për përdorimin efektiv të këtyre mjeteve nevojitet përvojë, dhe përvoja kërkon para. Unë jam i lumtur që kisha burime për të paguar, por shumë të tjerë që mund të bëjnë të njëjtën punë më mirë se unë, kurrë nuk do të kenë mundësinë për shkak të mungesës së fondeve edhe për një përpjekje.
Mjetet për të dhënat e mëdha janë universale. Nëse keni kohë, pothuajse me siguri do të mund të shkruani një zgjidhje më të shpejtë duke aplikuar pastrimin e të dhënave 'inteligjent', ruajtjen dhe metodikat e nxjerrjes. Në fund të fundit, gjithçka shkonte në analizën e kostove dhe përfitimeve.
Çfarë kam mësuar:
- nuk ka një mënyrë të lirë për të analizuar 25 TB të dhëna njëherësh;
- bëni kujdes me madhësinë e skedarëve tuaj Parquet dhe organizimin e tyre;
- particionet në Spark duhet të jenë të balancuara;
- kurrë mos u përpiqni të bëni 2.5 milion pjesë;
- organizimi ende është i vështirë, ashtu si konfigurimi i Spark;
- ndonjëherë, të dhënat e veçanta kërkojnë zgjidhje të veçanta;
- bashkimi në Spark funksionon shpejt, por pjesëtimi vazhdon të jetë i kushtushëm;
- mos flini kur ju mësojnë bazat, me siguri dikush e ka zgjidhur problemin tuaj që në vitet 1980;
gnu parallelështë një gjë magjike, të gjithë duhet ta përdorin;- Spark e do të dhënat e papërpunuara dhe nuk i pëlqen të kombinojë pjesët;
- në Spark ka shumë shpenzime të tepërta në zgjidhjen e detyrave të thjeshta;
- të dhënat asociative në AWK janë shumë efikase;
- mund të aksesoni
stdindhestdoutnga skenari R, kështu që mund ta përdorni në proces; - falë realizimit të mençur të rrugëve S3, mund të trajtojë shumë skedarë;
- arsyeja kryesore e humbjes së kohës është optimizimi i parakohshëm i metodës suaj të ruajtjes;
- mos u përpiqni të optimizoni detyrat manualisht, lejoni që kompjuteri ta bëjë këtë;
- API duhet të jetë i thjeshtë për thjeshtësinë dhe fleksibilitetin e përdorimit;
- nëse të dhënat tuaja janë mirë të përgatitura, do të jetë e lehtë të kesh në memorie!
Burimi: habr.com
