


Hur man läser den här artikeln: Jag ber om ursäkt för att texten är så lång och kaotisk. För att spara tid börjar jag varje kapitel med en introduktion "Vad jag lärde mig" som sammanfattar kapitlet i en eller två meningar.
"Visa mig bara lösningen!" Om du bara vill se var jag har hamnat, hoppa till kapitlet "Bli mer uppfinningsrik", men jag tycker att det är mer intressant och användbart att läsa om misslyckandena.
Jag fick nyligen i uppdrag att sätta upp en process för att bearbeta en stor volym av råa DNA-sekvenser (tekniskt sett ett SNP-chip). Det fanns ett behov av att snabbt få data om en given genetisk plats (kallad SNP) för efterföljande modellering och andra uppgifter. Med R och AWK kunde jag rengöra och organisera data på ett naturligt sätt, vilket avsevärt påskyndade frågebehandlingen. Detta var inte lätt för mig och krävde många upprepningar. Den här artikeln hjälper dig att undvika några av mina misstag och visar dig vad jag fick.
Först några inledande förklaringar.
Data
Vårt universitets centrum för behandling av genetisk information försåg oss med 25 Tb TSV-data. Jag fick dem uppdelade i 5 Gzip-komprimerade paket, som vart och ett innehöll cirka 240 2,5GB-filer. Varje rad innehöll data för en SNP från en individ. Totalt överfördes data om ~60 miljoner SNP:er och ~30 tusen människor. Utöver SNP-information innehöll filerna många kolumner med siffror som speglar olika egenskaper såsom läsintensitet, frekvens av olika alleler etc. Totalt fanns det cirka XNUMX kolumner med unika värden.
Mål
Som med alla datahanteringsprojekt var det viktigaste att bestämma hur datan skulle användas. I det här fallet vi kommer mestadels att välja modeller och arbetsflöden för SNP baserat på SNP. Det vill säga, åt gången kommer vi att behöva data på endast en SNP. Jag var tvungen att lära mig att extrahera alla poster relaterade till en av de 2,5 miljoner SNP:erna så enkelt, snabbt och billigt som möjligt.
Hur man inte gör detta
Låt mig citera en passande kliché:
Jag misslyckades inte tusen gånger, jag upptäckte bara tusen sätt att inte analysera en massa data till ett frågebart format.
Första försöket
Vad jag lärde mig: Det finns inget billigt sätt att analysera 25 TB på en gång.
Efter att ha tagit en kurs som heter "Advanced Big Data Processing" vid Vanderbilt University, var jag säker på att jag hade räknat ut det. Det kommer förmodligen att ta en timme eller två att ställa in Hive-servern för att köra igenom all data och rapportera resultaten. Eftersom vår data lagras i AWS S3 använde jag tjänsten , som låter dig tillämpa Hive SQL-frågor på S3-data. Det finns inget behov av att konfigurera/konfigurera ett Hive-kluster, och du betalar bara för den data du letar efter.
Efter att jag visade Athena mina data och dess format, körde jag några tester med frågor som detta:
select * from intensityData limit 10;Och fick snabbt välstrukturerade resultat. Redo.
Tills vi försökte använda data i vårt arbete...
Jag blev ombedd att dra ut all SNP-data för att testa modellen på. Jag körde frågan:
select * from intensityData
where snp = 'rs123456';...och började vänta. Efter åtta minuter och mer än 4 TB begärd data fick jag resultatet. Athena debiterar baserat på mängden data som den hittar, till $5 per terabyte. Så denna enda begäran kostade $20 och åtta minuters väntan. För att köra modellen på all data skulle vi ha behövt vänta 38 år och betala 50 miljoner dollar. Det var uppenbarligen inte rätt för oss.
Borde ha använt parkett...
Vad jag lärde mig: Var försiktig med storleken på dina Parquet-filer och deras organisation.
Först försökte jag fixa situationen genom att konvertera alla TSV till . De är bekväma för att arbeta med stora datamängder eftersom informationen i dem lagras i ett kolumnformat: varje kolumn lagras i sitt eget minne/disksegment, till skillnad från textfiler, där raderna innehåller elementen i varje kolumn. Och om du behöver hitta något behöver du bara läsa den nödvändiga spalten. Dessutom lagrar varje fil ett intervall av värden i en kolumn, så om värdet du letar efter inte är i kolumnens intervall, kommer Spark inte att slösa tid på att skanna hela filen.
Jag körde en enkel uppgift att konvertera våra TSV till Parkett och släppte de nya filerna i Athena. Det tog ca 5 timmar. Men när jag körde frågan tog det ungefär lika lång tid och lite mindre pengar att slutföra. Saken är den att Spark, i ett försök att optimera uppgiften, helt enkelt packade upp en TSV-bit och lade den i sin egen Parkett-bit. Och eftersom varje bit var tillräckligt stor för att innehålla de fullständiga uppgifterna för många människor, innehöll varje fil alla SNP:er, så Spark var tvungen att öppna alla filer för att extrahera den information den behövde.
Intressant nog är den förinställda (och rekommenderade) komprimeringstypen i Parkett, snappy, inte delbar. Därför var varje executor fast i uppgiften att packa upp och ladda hela 3,5 GB dataset.
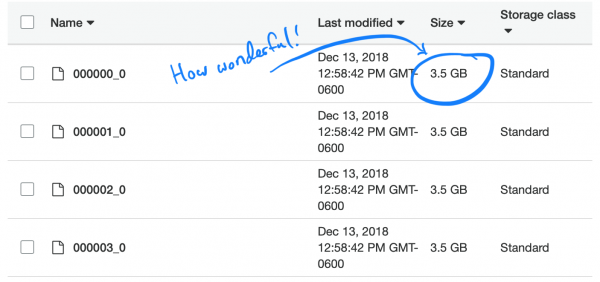
Låt oss undersöka problemet
Vad jag lärde mig: Sortering är svårt, speciellt om data är distribuerad.
Jag trodde att jag nu förstod kärnan i problemet. Jag behövde bara sortera data efter SNP-kolumnen, inte efter personer. Sedan kommer flera SNP:er att lagras i en separat dataklump, och då kommer Parquets "smarta" "öppna endast om värdet är inom intervallet"-funktion att visa sig i all ära. Tyvärr visade det sig vara en svår uppgift att sortera miljarder rader utspridda över klustret.
Jag går på algoritmkurs på college: "Usch, ingen bryr sig om beräkningskomplexiteten för alla dessa sorteringsalgoritmer"
Jag försöker sortera på en kolumn i en 20TB tabell: "Varför tar det här så lång tid?" kämpar.
— Nick Strayer (@NicholasStrayer)
AWS vill definitivt inte återbetala pengar eftersom "jag är en distraherad student." Efter att jag började sortera på Amazon Glue fungerade det i 2 dagar och kraschade sedan.
Hur är det med partitionering?
Vad jag lärde mig: Skiljeväggar i Spark måste vara balanserade.
Sedan kom jag på idén att dela upp data i kromosomer. Det finns 23 av dem (och några fler om man tar hänsyn till mitokondrie-DNA och omappade regioner).
Detta gör att du kan dela upp data i mindre bitar. Om du bara lägger till en rad till Spark-exportfunktionen i Glue-skriptet partition_by = "chr", då måste data distribueras i hinkar.
Genomet består av många fragment som kallas kromosomer.
Tyvärr fungerade det inte. Kromosomer har olika storlekar, och därför olika mängder information. Detta innebar att uppgifterna som Spark skickade till arbetarna var obalanserade och gick långsamt eftersom vissa noder slutade tidigt och var inaktiva. Uppgifterna var dock klara. Men när man bad om en enda SNP blev obalanser återigen ett problem. Kostnaden för att bearbeta SNP på större kromosomer (det vill säga där vi vill hämta data) har bara minskat med cirka 10 gånger. Mycket, men inte tillräckligt.
Vad händer om vi delar upp det i ännu mindre partitioner?
Vad jag lärde mig: Försök aldrig att göra 2,5 miljoner partitioner alls.
Jag bestämde mig för att gå all out och partitionera varje SNP. Detta säkerställde att partitionerna var lika stora. DET VAR EN DÅLIG IDÉ. Jag använde Glue och lade till en oskyldig linje partition_by = 'snp'. Uppgiften har startat och börjat utföras. En dag senare kollade jag och såg att inget skrivits till S3 ännu, så jag avbröt uppgiften. Det ser ut som att Glue skrev mellanfiler till en dold plats i S3, och många filer, kanske ett par miljoner. Som ett resultat av detta kostade mitt misstag mig över tusen dollar och behagade inte min mentor.
Partitionering + sortering
Vad jag lärde mig: Sortering är fortfarande svårt, liksom att trimma Spark.
Det sista försöket med att partitionera var att jag partitionerade kromosomerna och sedan sorterade varje partition. I teorin skulle detta påskynda varje fråga eftersom den önskade SNP-datan måste ligga inom några parkettbitar inom det givna intervallet. Tyvärr visade det sig vara en svår uppgift att sortera även partitionerade data. Som ett resultat bytte jag till EMR för ett anpassat kluster och använde åtta kraftfulla instanser (C5.4xl) och Sparklyr för att skapa ett mer flexibelt arbetsflöde...
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)… men uppgiften var fortfarande inte klar. Jag provade olika justeringar: ökade minnesallokering för varje frågeexekutor, använde noder med mer minne, använde sändningsvariabler, men varje gång visade det sig vara halvmått, och gradvis började executorerna misslyckas tills allt stannade.
Uppdatering: så börjar det.
— Nick Strayer (@NicholasStrayer)
Jag blir mer uppfinningsrik
Vad jag lärde mig: Ibland kräver specialdata speciallösningar.
Varje SNP har ett positionsvärde. Detta är ett tal som motsvarar antalet baser som ligger längs dess kromosom. Detta är ett bra och naturligt sätt att organisera vår data. Först ville jag dela upp efter regioner av varje kromosom. Till exempel positioner 1 - 2000, 2001 - 4000, etc. Men problemet är att SNP inte är jämnt fördelade över kromosomerna, så storleken på grupperna kommer att variera kraftigt.
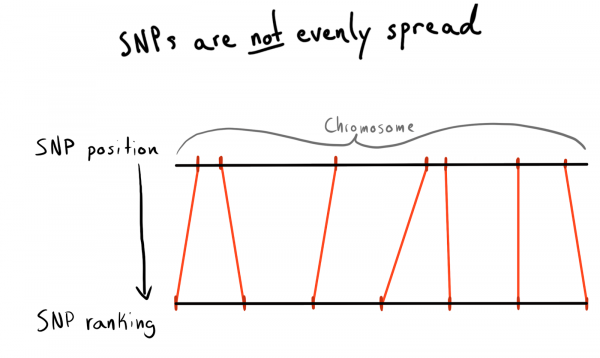
Som ett resultat kom jag till en uppdelning i kategorier (rang) av positioner. Baserat på redan laddade data, körde jag en fråga för att få en lista över unika SNP, deras positioner och kromosomer. Sedan sorterade jag data inom varje kromosom och samlade in SNP:erna i grupper (bins) av en given storlek. Låt oss säga 1000 SNP. Detta gav mig SNP-associationen med gruppen-på-kromosomen.
Till slut gjorde jag papperskorgar med 75 SNP, anledningen kommer att förklaras nedan.
snp_to_bin <- unique_snps %>%
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()Försök först med Spark
Vad jag lärde mig: Spark joins är snabba, men partitionering är fortfarande dyrt.
Jag ville läsa denna lilla (2,5 miljoner rader) dataram i Spark, slå samman den med rådata och sedan partitionera efter den nyligen tillagda kolumnen bin.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
)
jag använde sdf_broadcast(), så Spark vet att den behöver skicka dataramen till alla noder. Detta är användbart om informationen är liten och krävs för alla uppgifter. Annars försöker Spark vara smart och distribuerar data efter behov, vilket kan orsaka nedgångar.
Och återigen fungerade inte min idé: uppgifterna fungerade under en tid, slutförde sammanslagningen, och sedan började de misslyckas, precis som exekutörerna som startade genom partitionering.
Lägger till AWK
Vad jag lärde mig: Sov inte när du lär dig grunderna. Någon har säkert löst ditt problem redan på 1980-talet.
Fram till denna punkt orsakades alla mina misslyckanden med Spark av att data blandades ihop i klustret. Kanske kan situationen förbättras med förbehandling. Jag bestämde mig för att försöka dela upp den råa textdatan i kromosomkolumner, som jag hoppades skulle förse Spark med "förpartitionerade" data.
Jag sökte på StackOverflow hur man delar upp efter kolumnvärden och hittade Med AWK kan du dela upp en textfil efter kolumnvärden genom att skriva till ett skript istället för att skicka resultaten till stdout.
För att testa detta skrev jag ett Bash-manus. Laddade ner en av de packade TSV:erna och packade sedan upp den med gzip och skickas till awk.
gzip -dc path/to/chunk/file.gz |
awk -F 't'
'{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'Det fungerade!
Fyller kärnorna
Vad jag lärde mig: gnu parallel – Det är en magisk grej, alla borde använda den.
Separationen var ganska långsam och när jag började htopför att testa användningen av en kraftfull (och dyr) EC2-instans visar det sig att jag bara använder en kärna och cirka 200 MB minne. För att lösa problemet och inte förlora massor av pengar var vi tvungna att ta reda på hur vi skulle parallellisera arbetet. Som tur är i en helt fantastisk bok Jag hittade ett kapitel om parallellism av Jeroen Janssens. Av det lärde jag mig om gnu parallel, en mycket flexibel metod för att implementera multithreading i Unix.

När jag började splittra med den nya processen var allt bra, men det fanns fortfarande en flaskhals - att ladda ner S3-objekt till disk var inte särskilt snabb och inte helt parallelliserad. För att fixa detta gjorde jag följande:
- Jag fick reda på att det är möjligt att implementera S3-nedladdningssteget direkt i pipeline, vilket helt eliminerar mellanlagring på disk. Det betyder att jag kan undvika att skriva rådata till disk och använda ännu mindre, och därför billigare, lagring på AWS.
- Team
aws configure set default.s3.max_concurrent_requests 50ökade kraftigt antalet trådar som AWS CLI använder (standard är 10). - Bytte till en nätverksoptimerad EC2-instans med bokstaven n i namnet. Jag fann att förlusten i processorkraft vid användning av n-instanser mer än kompenseras av ökningen i laddningshastighet. För de flesta uppgifter använde jag c5n.4xl.
- Ändrad
gzippå , är ett gzip-verktyg som kan göra coola saker för att parallellisera den initialt icke-parallella uppgiften att packa upp filer (detta hjälpte minst).
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe
"awk -F 't' '{print $1",..."$30">"chunked/{#}_chr"$15".csv"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneDessa steg kombineras med varandra för att få allt att fungera väldigt snabbt. Genom att öka nedladdningshastigheterna och eliminera diskskrivningar kunde jag nu bearbeta en 5 terabyte batch på bara några timmar.
Det finns inget sötare än att se alla kärnor du betalar för på AWS används. Tack vare gnu-parallel kan jag packa upp och dela upp en 19 gig csv lika snabbt som jag kan ladda ner den. Jag kunde inte ens få gnista för att köra detta.
— Nick Strayer (@NicholasStrayer)
Den här tweeten var tänkt att nämna "TSV". Tyvärr.
Använder omtolkad data
Vad jag lärde mig: Spark gillar okomprimerad data och gillar inte att kombinera partitioner.
Nu fanns data i S3 i ett okomprimerat (läs: delat) och halvordnat format, och jag kunde gå tillbaka till Spark. En överraskning väntade mig: jag lyckades inte uppnå det jag ville igen! Det var mycket svårt att berätta för Spark exakt hur data var uppdelad. Och även när jag gjorde detta visade det sig att det fanns för många partitioner (95 tusen), och när jag använde coalesce Jag minskade deras antal till rimliga gränser, detta förstörde min partitionering. Jag är säker på att detta går att fixa, men efter ett par dagars letande kunde jag inte hitta någon lösning. Jag avslutade så småningom alla uppgifter i Spark, även om det tog lite tid och mina delade parkettfiler var inte särskilt små (~200 KB). Datan var dock där den behövde vara.
För litet och annorlunda, underbart!
Testa lokala gnistfrågor
Vad jag lärde mig: Spark har för mycket omkostnader när man löser enkla problem.
Genom att ladda ner data i ett väldesignat format kunde jag testa hastigheten. Konfigurerade ett R-skript för att starta en lokal Spark-server och laddade sedan en Spark-dataram från den angivna Parquet-grupplagringen (bin). Jag försökte ladda all data men kunde inte få Sparklir att känna igen partitioneringen.
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Start a timer
start_time <- Sys.time()
# Load the desired bin into Spark
intensity_data <- sc %>%
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Subset bin to snp and then collect to local
test_subset <- intensity_data %>%
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)Avrättningen tog 29,415 sekunder. Mycket bättre, men inte särskilt bra för masstestning av någonting. Jag kunde inte heller snabba upp saker och ting med cachelagring eftersom när jag försökte cachelagra en dataram i minnet kraschade Spark alltid, även när jag tilldelade mer än 50 GB minne för en datauppsättning som var mindre än 15 GB.
Återgå till AWK
Vad jag lärde mig: Associativa arrayer i AWK är mycket effektiva.
Jag visste att jag kunde uppnå högre hastighet. Jag kom ihåg det i en underbar Jag läste om en cool funktion som heter "". I grund och botten är dessa nyckel-värdepar, som av någon anledning hette annorlunda i AWK, så jag tänkte på något sätt inte riktigt på dem. påminde om att termen "associativa arrayer" är mycket äldre än termen "nyckel-värdepar". Även om du , du kommer inte att se denna term där, men du kommer att hitta associativa arrayer! Dessutom är "nyckel-värdepar" oftast associerat med databaser, så det är mycket mer meningsfullt att jämföra det med hashmap. Jag insåg att jag kunde använda dessa associativa arrayer för att länka mina SNP till bin-tabellen och rådata utan att använda Spark.
För detta ändamål använde jag ett block i AWK-skriptet BEGIN. Detta är en bit kod som exekveras innan den första raden med data skickas till skriptets huvuddel.
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline < "snp_to_bin.csv") {bin[$1] = $2}
}
{
print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
}
Team while(getline...) laddade alla rader från CSV-gruppen (bin), ange första kolumnen (SNP-namn) som nyckel för associativ array bin och det andra värdet (gruppen) som värdet. Sedan i blocket { }, som exekveras mot alla rader i huvudfilen, skickas varje rad till utdatafilen, som får ett unikt namn beroende på dess grupp (bin): ..._bin_"bin[$1]"_....
variabler batch_num и chunk_id matchade data som tillhandahålls av pipeline, vilket undviker tävlingsförhållanden, och varje utförandetråd startade parallel, skrev i sin egen unika fil.
Eftersom jag hade all rådata utspridda över mappar per kromosom kvar från mitt tidigare AWK-experiment, kunde jag nu skriva ett annat Bash-skript för att bearbeta det en kromosom i taget och mata de djupare partitionerade data till S3.
DESIRED_CHR='13'
# Download chromosome data from s3 and split into bins
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'reading {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=""$DESIRED_CHR"" -v chunk="{}" -f split_on_chr_bin.awk"
# Combine all the parallel process chunks to single files and upload to rds using R
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/*
Manuset har två avsnitt parallel.
I det första avsnittet läses data från alla filer som innehåller information om den erforderliga kromosomen, sedan distribueras dessa data mellan strömmar som distribuerar filer till lämpliga grupper (bin). För att undvika tävlingsförhållanden när flera trådar skriver till samma fil, skickar AWK filnamn för att skriva data till olika platser, till exempel, chr_10_bin_52_batch_2_aa.csv. Som ett resultat skapas många små filer på disken (för detta använde jag terabyte EBS-volymer).
Transportör från andra sektionen parallel går igenom grupper (bin) och slår samman sina individuella filer till gemensamma CSV c catoch skickar dem sedan för export.
Översättning till R?
Vad jag lärde mig: du kan kontakta stdin и stdout från ett R-skript, och använd det därför i pipelinen.
I Bash-skriptet kanske du har märkt en rad så här: ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... Den översätter alla sammanlänkade filer i en grupp (bin) till R-skriptet nedan. {} är en speciell teknik parallel, som infogar all data som skickas av den till den angivna strömmen direkt i själva kommandot. Alternativ {#} ger ett unikt tråd-ID och {%} representerar uppgiftsluckans nummer (upprepas, men aldrig samtidigt). En lista över alla alternativ finns i
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
)
När variabeln file("stdin") överförs till readr::read_csv, laddas data som översatts till R-skriptet in i en ram, som sedan konverteras till .rds-fil med hjälp av aws.s3 skrivet direkt till S3.
RDS är ungefär en juniorversion av Parkett, utan krusidullerna av en pelarförvaring.
Efter att Bash-manuset var klart fick jag ett gäng .rds-filer lagrade i S3, vilket gjorde att jag kunde använda effektiv komprimering och inbyggda typer.
Trots att man använde bromsen R fungerade allt väldigt snabbt. Det är inte förvånande att de delar av R som ansvarar för att läsa och skriva data är väl optimerade. Efter testning på en enda medelstor kromosom slutfördes jobbet på en C5n.4xl-instans på cirka två timmar.
S3 Begränsningar
Vad jag lärde mig: Tack vare smart implementering av sökvägar kan S3 hantera många filer.
Jag var orolig om S3 skulle kunna hantera det stora antalet filer som överfördes till den. Jag skulle kunna göra filnamnen meningsfulla, men hur skulle S3 söka efter dem?

Mappar i S3 är bara för att visa, systemet bryr sig inte riktigt om symbolen /.
Det verkar som att S3 representerar sökvägen till en specifik fil som en enkel nyckel i någon sorts dokumentbaserad hashtabell eller databas. En hink kan ses som en tabell och filer som poster i den tabellen.
Eftersom snabbhet och effektivitet är viktiga för att tjäna pengar på Amazon är det ingen överraskning att detta nyckel-som-fil-sökvägssystem är otroligt optimerat. Jag försökte hitta en balans mellan att inte behöva göra många get-förfrågningar, men ändå göra förfrågningarna snabba. Det visade sig att det är bäst att göra cirka 20 tusen bin-filer. Jag tror att om vi fortsätter att optimera kan vi uppnå hastighetsförbättringar (till exempel genom att göra en speciell hink enbart för data, och därmed minska storleken på uppslagstabellen). Men det fanns varken tid eller pengar för ytterligare experiment.
Hur är det med korskompatibilitet?
Vad jag har lärt mig: Den främsta orsaken till bortkastad tid är för tidig optimering av din lagringsmetod.
Vid det här laget är det mycket viktigt att fråga dig själv: "Varför använda ett proprietärt filformat?" Anledningen ligger i laddningshastigheten (gzip-packade CSV-filer tog 7 gånger längre tid att ladda) och kompatibilitet med våra arbetsflöden. Jag kan ompröva mitt beslut om R enkelt kan ladda Parquet (eller Arrow) filer utan bördan av Spark. I vårt labb använder alla R, och om jag behöver omvandla data till ett annat format har jag fortfarande den ursprungliga textdatan, så jag kan bara köra pipelinen igen.
Arbetsfördelning
Vad jag lärde mig: Försök inte att optimera uppgifter manuellt, låt datorn göra det.
Jag har felsökt arbetsflödet på en kromosom, nu måste jag bearbeta alla andra data.
Jag ville spinna upp flera EC2-instanser för konverteringen, men samtidigt var jag försiktig med att få en mycket obalanserad belastning över olika bearbetningsjobb (precis som Spark led av obalanserade partitioner). Dessutom kände jag inte för att köra en instans per kromosom eftersom AWS-konton har en standardgräns på 10 instanser.
Sedan bestämde jag mig för att skriva ett manus i R för att optimera bearbetningsuppgifter.
Först bad jag S3 att beräkna hur mycket lagringsutrymme varje kromosom tar upp.
library(aws.s3)
library(tidyverse)
chr_sizes <- get_bucket_df(
bucket = '...', prefix = '...', max = Inf
) %>%
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Extract chromosome from the file name
chr = str_extract(Key, 'chr.{1,4}.csv') %>%
str_remove_all('chr|.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Divide to get value in GB
# A tibble: 27 x 2
chr total_size
<chr> <dbl>
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … with 17 more rows
Sedan skrev jag en funktion som tar den totala storleken, blandar ordningen på kromosomerna och delar upp dem i grupper num_jobs och rapporterar hur mycket storleken på alla bearbetningsjobb skiljer sig åt.
num_jobs <- 7
# How big would each job be if perfectly split?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job <- function(i){
chr_sizes %>%
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)
# A tibble: 1 x 2
sd data
<dbl> <list>
1 153. <tibble [7 × 3]>Sedan körde jag tusen shuffles med purrr och valde den bästa.
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1)
Så jag fick en uppsättning uppgifter som var väldigt lika i storlek. Sedan var det bara att linda in mitt tidigare Bash-manus i en stor loop for. Det tog cirka 10 minuter att skriva denna optimering. Och det är mycket mindre än jag skulle ha spenderat på att manuellt skapa uppgifter om de var obalanserade. Därför tror jag att jag tog rätt beslut med denna preliminära optimering.
for DESIRED_CHR in "16" "9" "7" "21" "MT"
do
# Code for processing a single chromosome
fiI slutet lägger jag till kommandot shutdown:
sudo shutdown -h now
...och allt löste sig! Jag använde AWS CLI för att starta instanser och genom alternativet user_data skickade dem Bash-skript av deras uppgifter för bearbetning. De körde och stängdes av automatiskt, så jag betalade inte för extra datorkraft.
aws ec2 run-instances ...
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]"
--user-data file://<<job_script_loc>>Låt oss packa!
Vad jag lärde mig: API:et bör vara enkelt för enkelhetens och flexibilitetens skull.
Äntligen fick jag uppgifterna på rätt plats och i rätt form. Allt som återstod var att förenkla processen med att använda data så mycket som möjligt för att underlätta för mina kollegor. Jag ville göra ett enkelt API för att göra förfrågningar. Om jag i framtiden bestämmer mig för att byta från .rds på Parkettfiler, så borde detta vara ett problem för mig, inte för mina kollegor. För detta bestämde jag mig för att göra ett internt R-paket.
Jag satte ihop och dokumenterade ett väldigt enkelt paket som bara innehåller några få dataåtkomstfunktioner, organiserat runt funktionen get_snp. Jag gjorde även en hemsida för mina kollegor så att de enkelt kan titta på exempel och dokumentation.
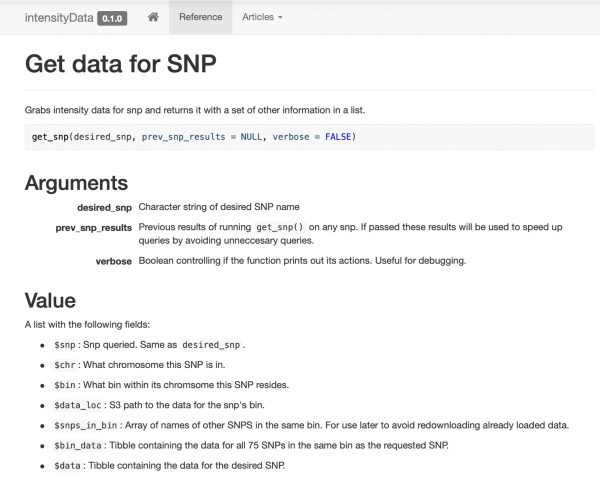
Intelligent cachelagring
Vad jag lärde mig: Om din data är väl förberedd blir caching enkelt!
Eftersom ett av huvudarbetsflödena var att tillämpa samma analysmodell på en grupp SNP:er, bestämde jag mig för att använda binning till min fördel. Vid överföring av data via SNP bifogas all information från gruppen (bin) till det returnerade objektet. Det vill säga att gamla förfrågningar kan (i teorin) påskynda behandlingen av nya förfrågningar.
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
...
När jag byggde paketet körde jag många riktmärken för att jämföra hastigheten på olika metoder. Jag rekommenderar att inte försumma detta, för ibland är resultaten oväntade. Till exempel, dplyr::filter var mycket snabbare än att ta tag i rader med indexbaserad filtrering, och att få en enda kolumn från en filtrerad dataram var mycket snabbare än att använda indexeringssyntaxen.
Observera att objektet prev_snp_results innehåller nyckeln snps_in_bin. Detta är en uppsättning av alla unika SNP:er i en grupp (bin), så att du snabbt kan kontrollera om det redan finns data från en tidigare fråga. Det gör det också enkelt att gå igenom alla SNP:er i en fack med denna kod:
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}Resultat
Vi kan nu (och har börjat på allvar) köra modeller och scenarier som tidigare var otillgängliga för oss. Det bästa är att mina labbkollegor inte behöver tänka på någon av komplexiteten. De har bara en funktion som fungerar.
Och medan paketet besparar dem detaljerna, försökte jag göra dataformatet så enkelt att de kunde förstå det om jag plötsligt försvann imorgon...
Hastigheten har ökat märkbart. Vanligtvis skannar vi funktionellt signifikanta fragment av genomet. Vi kunde inte göra detta tidigare (det var för dyrt), men nu, tack vare binstrukturen och cachningen, tar det mindre än 0,1 sekunder i genomsnitt att fråga en enskild SNP, och dataanvändningen är så låg att S3-kostnaderna är nästan ingenting.
Nyligen fick jag en förändring av att gräla över 25+ TB av rå genotypningsdata för mitt labb. När jag började tog det 8 minuter att använda spark och kostade $20 för att fråga en SNP. Efter att ha använt AWK + att bearbeta tar det nu mindre än en tiondels sekund och kostar $10. Min personliga vinna.
— Nick Strayer (@NicholasStrayer)
Slutsats
Den här artikeln är inte alls en guide. Lösningen visade sig vara individuell, och nästan säkert inte optimal. Det är snarare en berättelse om en resa. Jag vill att andra ska förstå att beslut som dessa inte bara dyker upp i ditt huvud fullt utformade, de är resultatet av försök och misstag. Dessutom, om du letar efter en datavetare, kom ihåg att användningen av dessa verktyg effektivt kräver erfarenhet och erfarenhet kostar pengar. Jag är glad att jag hade möjlighet att betala, men många andra som kan göra samma jobb bättre än jag kommer aldrig att få möjlighet på grund av brist på pengar att ens prova.
Big data-verktyg är mångsidiga. Om du har tid kan du nästan säkert skriva en snabbare lösning med smarta tekniker för datarensning, lagring och extraktion. I slutändan handlar det om en kostnads-nyttoanalys.
Vad jag lärde mig:
- det finns inget billigt sätt att analysera 25 TB på en gång;
- var försiktig med storleken på dina Parquet-filer och deras organisation;
- partitioner i Spark måste vara balanserade;
- försök aldrig göra 2,5 miljoner partitioner alls;
- Sortering är fortfarande svårt, liksom att sätta upp Spark;
- ibland kräver speciella data speciella lösningar;
- Spark joins är snabba, men partitionering är fortfarande dyrt;
- sov inte när du lär dig grunderna, någon har förmodligen löst ditt problem redan på 1980-talet;
gnu parallel– det är en magisk sak, alla borde använda den;- Spark gillar okomprimerad data och gillar inte att kombinera partitioner;
- Spark har för mycket omkostnader när man löser enkla problem;
- associativa arrayer i AWK är mycket effektiva;
- du kan kontakta
stdinиstdoutfrån ett R-skript, och använd det därför i pipelinen; - Tack vare smart implementering av sökvägar kan S3 hantera många filer;
- Den främsta anledningen till att slösa tid är för tidig optimering av din lagringsmetod;
- försök inte att optimera uppgifter manuellt, låt datorn göra det;
- API:et bör vara enkelt för enkelhetens och flexibilitetens skull;
- Om din data är väl förberedd blir caching enkelt!
Källa: will.com
