


Ang kasalukuyang pandemya ng COVID-19 ay lumikha ng maraming problema na ikinatutuwang atakehin ng mga hacker. Mula sa 3D printed face shields at homemade medical masks hanggang sa pagpapalit ng full mechanical ventilator, ang daloy ng mga ideya ay nagbibigay inspirasyon at nakakapagpainit ng puso. Kasabay nito, may mga pagtatangka na sumulong sa ibang lugar: sa pananaliksik na naglalayong labanan ang virus mismo.
Tila, ang pinakamalaking potensyal para sa pagpapahinto sa kasalukuyang pandemya at pag-iwas sa lahat ng mga kasunod ay nakasalalay sa isang diskarte na sumusubok na makarating sa pinaka-ugat ng problema. Ang diskarteng ito na "kilalanin ang iyong kaaway" ay kinukuha ng proyekto ng Folding@Home computing. Milyun-milyong tao ang nag-sign up sa proyekto at nag-donate ng ilan sa kapangyarihan sa pagproseso ng kanilang mga processor at GPU, kaya lumilikha ng pinakamalaking [naipamahagi] supercomputer sa kasaysayan.
Ngunit para saan ba talaga ginagamit ang lahat ng mga exaflop na ito? Bakit kailangang itapon ang naturang computing power sa ? Anong uri ng biochemistry ang gumagana dito, bakit kailangang tupi ang mga protina? Narito ang isang mabilis na pangkalahatang-ideya ng pagtitiklop ng protina: ano ito, paano ito nangyayari, at bakit ito mahalaga.
Una, ang pinakamahalagang bagay: bakit kailangan ang mga protina?
Ang mga protina ay mahahalagang istruktura. Hindi lamang sila nagbibigay ng materyal na gusali para sa mga selula, ngunit nagsisilbi rin bilang mga katalista ng enzyme para sa halos lahat ng mga reaksiyong biochemical. Mga ardilya, maging sila o , ay mahahabang tanikala , na matatagpuan sa isang tiyak na pagkakasunud-sunod. Ang mga pag-andar ng mga protina ay tinutukoy kung aling mga amino acid ang matatagpuan sa ilang mga lugar sa protina. Kung, halimbawa, ang isang protina ay kailangang magbigkis sa isang positibong sisingilin na molekula, ang binding site ay dapat mapuno ng negatibong sisingilin na mga amino acid.
Upang maunawaan kung paano nakukuha ng mga protina ang istraktura na tumutukoy sa kanilang paggana, kailangan nating suriin ang mga pangunahing kaalaman ng molecular biology at ang daloy ng impormasyon sa cell.
Produksyon, o Ang mga protina ay nagsisimula sa proseso . Sa panahon ng transkripsyon, ang DNA double helix, na naglalaman ng genetic na impormasyon ng cell, ay bahagyang humiwalay, na nagpapahintulot sa mga nitrogen base ng DNA na maging available sa isang enzyme na tinatawag na . Ang trabaho ng RNA polymerase ay gumawa ng RNA copy, o transcription, ng isang gene. Ang kopyang ito ng isang gene na tinatawag na (mRNA), ay isang solong molekula na perpekto para sa pagkontrol sa mga pabrika ng protina ng intracellular, na nakikibahagi sa produksyon, o mga protina.
Ang mga ribosome ay kumikilos tulad ng mga makina ng pagpupulong - kinukuha nila ang template ng mRNA at itinutugma ito sa iba pang maliliit na piraso ng RNA, (tRNA). Ang bawat tRNA ay may dalawang aktibong rehiyon - isang seksyon ng tatlong base na tinatawag , na dapat tumugma sa kaukulang mga codon ng mRNA, at isang site para sa pagbubuklod ng isang amino acid na tiyak para dito . Sa panahon ng pagsasalin, ang mga molekula ng tRNA sa ribosome ay random na sumusubok na magbigkis sa mRNA gamit ang mga anticodon. Kung matagumpay, ikinakabit ng molekula ng tRNA ang amino acid nito sa nauna, na bumubuo ng susunod na link sa chain ng mga amino acid na naka-encode ng mRNA.
Ang pagkakasunud-sunod ng mga amino acid na ito ay ang unang antas ng hierarchy ng istruktura ng protina, kaya naman tinawag itong . Ang buong three-dimensional na istraktura ng isang protina at ang mga pag-andar nito ay direktang nagmula sa pangunahing istraktura, at nakasalalay sa iba't ibang katangian ng bawat isa sa mga amino acid at ang kanilang mga pakikipag-ugnayan sa isa't isa. Kung wala itong mga kemikal na katangian at pakikipag-ugnayan ng amino acid, mananatili silang mga linear sequence na walang three-dimensional na istraktura. Ito ay makikita sa tuwing nagluluto ka ng pagkain - sa prosesong ito ay may thermal tatlong-dimensional na istraktura ng mga protina.
Long-range na mga bono ng mga bahagi ng protina
Ang susunod na antas ng three-dimensional na istraktura, na lampas sa pangunahing isa, ay binigyan ng isang matalinong pangalan . Kabilang dito ang mga bono ng hydrogen sa pagitan ng mga amino acid na medyo malapit ang pagkilos. Ang pangunahing diwa ng mga nagpapatatag na pakikipag-ugnayan na ito ay bumaba sa dalawang bagay: и . Ang alpha helix ay bumubuo sa mahigpit na nakapulupot na rehiyon ng polypeptide, habang ang beta sheet ay bumubuo sa makinis at malawak na rehiyon. Ang parehong mga pormasyon ay may parehong istruktura at functional na mga katangian, depende sa mga katangian ng kanilang mga constituent amino acids. Halimbawa, kung ang alpha helix ay pangunahing binubuo ng mga hydrophilic amino acid, tulad ng o , pagkatapos ay malamang na lalahok ito sa mga may tubig na reaksyon.
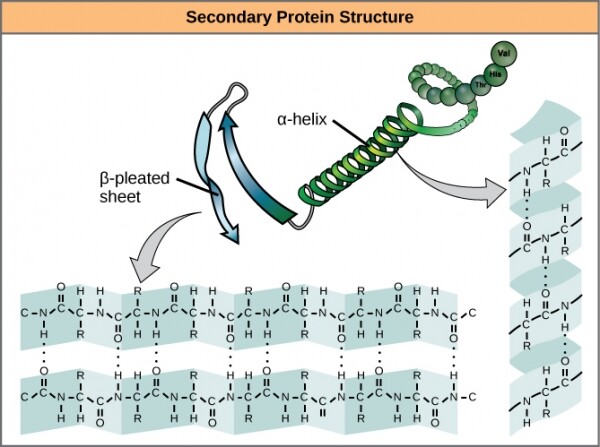
Mga alpha helice at beta sheet sa mga protina. Ang mga hydrogen bond ay nabuo sa panahon ng pagpapahayag ng protina.
Ang dalawang istrukturang ito at ang kanilang mga kumbinasyon ay bumubuo sa susunod na antas ng istraktura ng protina - . Hindi tulad ng mga simpleng fragment ng pangalawang istraktura, ang tertiary na istraktura ay pangunahing naiimpluwensyahan ng hydrophobicity. Ang mga sentro ng karamihan sa mga protina ay naglalaman ng mataas na hydrophobic amino acid, tulad ng o , at ang tubig ay hindi kasama doon dahil sa "mamantika" na katangian ng mga radikal. Ang mga istrukturang ito ay madalas na lumilitaw sa mga transmembrane na protina na naka-embed sa lipid bilayer membrane na nakapalibot sa mga cell. Ang mga hydrophobic na rehiyon ng mga protina ay nananatiling thermodynamically stable sa loob ng mataba na bahagi ng lamad, habang ang mga hydrophilic na rehiyon ng protina ay nakalantad sa may tubig na kapaligiran sa magkabilang panig.
Gayundin, ang katatagan ng mga istrukturang tersiyaryo ay sinisiguro ng mga pangmatagalang mga bono sa pagitan ng mga amino acid. Ang isang klasikong halimbawa ng gayong mga koneksyon ay , kadalasang nangyayari sa pagitan ng dalawang cysteine radicals. Kung naamoy mo ang isang bagay na medyo tulad ng mga bulok na itlog sa isang hair salon sa panahon ng isang perm procedure sa buhok ng isang kliyente, kung gayon ito ay isang bahagyang denaturation ng tertiary na istraktura ng keratin na nakapaloob sa buhok, na nangyayari sa pamamagitan ng pagbawas ng mga disulfide bond na may tulong ng sulfur-containing pinaghalong.
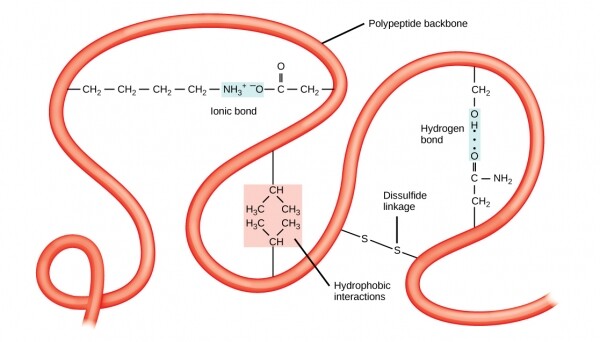
Ang tersiyaryong istraktura ay nagpapatatag sa pamamagitan ng pangmatagalang pakikipag-ugnayan tulad ng hydrophobicity o disulfide bond
Ang mga bono ng disulfide ay maaaring mangyari sa pagitan radical sa parehong polypeptide chain, o sa pagitan ng mga cysteine mula sa iba't ibang kumpletong chain. Nabubuo ang mga pakikipag-ugnayan sa pagitan ng iba't ibang chain antas ng istraktura ng protina. Ang isang mahusay na halimbawa ng quaternary na istraktura ay nasa dugo mo yan. Ang bawat molekula ng hemoglobin ay binubuo ng apat na magkaparehong globin, mga bahagi ng protina, na ang bawat isa ay hawak sa isang tiyak na posisyon sa loob ng polypeptide ng mga tulay na disulfide, at nauugnay din sa isang molekula ng heme na naglalaman ng bakal. Ang lahat ng apat na globin ay konektado sa pamamagitan ng intermolecular disulfide bridges, at ang buong molekula ay nagbubuklod sa ilang molekula ng hangin nang sabay-sabay, hanggang apat, at nagagawang palabasin ang mga ito kung kinakailangan.
Pagmomodelo ng mga istruktura sa paghahanap ng lunas para sa sakit
Nagsisimulang matiklop ang mga polypeptide chain sa kanilang huling hugis sa panahon ng pagsasalin, habang ang lumalagong chain ay lumalabas sa ribosome, katulad ng isang piraso ng memory-alloy wire na maaaring magkaroon ng mga kumplikadong hugis kapag pinainit. Gayunpaman, gaya ng nakasanayan sa biology, ang mga bagay ay hindi ganoon kadali.
Sa maraming mga cell, ang mga na-transcribe na gene ay sumasailalim sa malawak na pag-edit bago ang pagsasalin, na makabuluhang nagbabago sa pangunahing istraktura ng protina kumpara sa purong base sequence ng gene. Sa kasong ito, ang mga mekanismo ng pagsasalin ay madalas na humihingi ng tulong ng mga molecular chaperone, mga protina na pansamantalang nagbubuklod sa nascent polypeptide chain at pinipigilan itong kumuha ng anumang intermediate form, kung saan hindi na sila makakapatuloy sa pangwakas.
Ito ay upang sabihin na ang paghula sa huling hugis ng isang protina ay hindi isang maliit na gawain. Sa loob ng mga dekada, ang tanging paraan upang pag-aralan ang istruktura ng mga protina ay sa pamamagitan ng mga pisikal na pamamaraan tulad ng X-ray crystallography. Ito ay hindi hanggang sa huling bahagi ng 1960s na ang mga biophysical chemist ay nagsimulang bumuo ng mga computational na modelo ng pagtitiklop ng protina, pangunahing nakatuon sa pangalawang pagmomolde ng istraktura. Ang mga pamamaraang ito at ang mga inapo ng mga ito ay nangangailangan ng napakalaking halaga ng input data bilang karagdagan sa pangunahing istraktura - halimbawa, mga talahanayan ng mga anggulo ng bono ng amino acid, mga listahan ng hydrophobicity, mga naka-charge na estado, at maging ang konserbasyon ng istraktura at paggana sa mga evolutionary timescale - lahat upang hulaan kung ano ang mangyayari mukhang ang huling protina.
Ang mga computational na pamamaraan ngayon para sa pangalawang paghula ng istraktura, tulad ng mga tumatakbo sa Folding@Home network, ay gumagana nang may humigit-kumulang 80% na katumpakan—na medyo maganda kung isasaalang-alang ang pagiging kumplikado ng problema. Ang data na nabuo ng mga predictive na modelo sa mga protina tulad ng SARS-CoV-2 spike protein ay ihahambing sa data mula sa mga pisikal na pag-aaral ng virus. Bilang resulta, posible na makuha ang eksaktong istraktura ng protina at, marahil, maunawaan kung paano nakakabit ang virus sa mga receptor. isang tao na matatagpuan sa respiratory tract na humahantong sa katawan. Kung malalaman natin ang istrukturang ito, maaari tayong makahanap ng mga gamot na humaharang sa pagbubuklod at maiwasan ang impeksiyon.
Ang pananaliksik sa pagtitiklop ng protina ay nasa puso ng aming pag-unawa sa napakaraming sakit at impeksyon na kahit na ginagamit namin ang Folding@Home network upang malaman kung paano talunin ang COVID-19, na nakita naming sumabog sa paglago kamakailan, nanalo ang network. t maging idle nang matagal. trabaho. Ito ay isang tool sa pagsasaliksik na angkop para sa pag-aaral ng mga pattern ng protina na sumasailalim sa dose-dosenang mga sakit na nauugnay sa misfolding ng protina, tulad ng Alzheimer's disease o isang variant ng Creutzfeldt-Jakob disease, na kadalasang hindi wastong tinatawag na mad cow disease. At kapag ang isa pang virus ay hindi maiiwasang lumitaw, kami ay magiging handa upang simulan muli itong labanan.
Pinagmulan: www.habr.com
