


اس مضمون کو کیسے پڑھیں: متن کے اتنے لمبے اور افراتفری کے لیے معذرت خواہ ہوں۔ آپ کا وقت بچانے کے لیے، میں ہر باب کا آغاز "میں نے کیا سیکھا" کے تعارف سے کیا ہے، جو ایک یا دو جملوں میں باب کے جوہر کا خلاصہ کرتا ہے۔
"بس مجھے حل دکھائیں!" اگر آپ صرف یہ دیکھنا چاہتے ہیں کہ میں کہاں سے آیا ہوں، تو باب "زیادہ اختراعی بننا" پر جائیں، لیکن میرے خیال میں ناکامی کے بارے میں پڑھنا زیادہ دلچسپ اور مفید ہے۔
مجھے حال ہی میں خام ڈی این اے کی ترتیب کی ایک بڑی مقدار (تکنیکی طور پر ایک SNP چپ) پر کارروائی کے لیے ایک عمل ترتیب دینے کا کام سونپا گیا تھا۔ ضرورت اس بات کی تھی کہ بعد میں ماڈلنگ اور دیگر کاموں کے لیے دیے گئے جینیاتی مقام (جسے SNP کہا جاتا ہے) کے بارے میں فوری طور پر ڈیٹا حاصل کیا جائے۔ R اور AWK کا استعمال کرتے ہوئے، میں ڈیٹا کو قدرتی طریقے سے صاف اور منظم کرنے کے قابل تھا، جس سے استفسار کی کارروائی میں تیزی آئی۔ یہ میرے لیے آسان نہیں تھا اور متعدد تکرار کی ضرورت تھی۔ یہ مضمون آپ کو میری کچھ غلطیوں سے بچنے میں مدد دے گا اور آپ کو دکھائے گا کہ میں نے کیا کیا۔
سب سے پہلے، کچھ تعارفی وضاحتیں.
ڈیٹا
ہمارے یونیورسٹی کے جینیٹک انفارمیشن پروسیسنگ سینٹر نے ہمیں 25 TB TSV کی شکل میں ڈیٹا فراہم کیا۔ میں نے انہیں 5 پیکجوں میں تقسیم کیا، Gzip کے ذریعے کمپریس کیا گیا، جن میں سے ہر ایک میں تقریباً 240 چار گیگا بائٹ فائلیں تھیں۔ ہر قطار میں ایک فرد سے ایک SNP کا ڈیٹا ہوتا ہے۔ مجموعی طور پر، 2,5 ملین SNPs اور ~ 60 ہزار لوگوں کا ڈیٹا منتقل کیا گیا۔ SNP کی معلومات کے علاوہ، فائلوں میں متعدد کالم ہوتے ہیں جن میں مختلف خصوصیات کی عکاسی ہوتی ہے، جیسے پڑھنے کی شدت، مختلف ایللیس کی فریکوئنسی وغیرہ۔ مجموعی طور پر منفرد اقدار کے ساتھ تقریباً 30 کالم تھے۔
گول
کسی بھی ڈیٹا مینجمنٹ پروجیکٹ کی طرح، سب سے اہم چیز یہ طے کرنا تھی کہ ڈیٹا کو کس طرح استعمال کیا جائے گا۔ اس معاملے میں ہم SNP کی بنیاد پر SNP کے لیے زیادہ تر ماڈلز اور ورک فلو کا انتخاب کریں گے۔. یعنی، ہمیں ایک وقت میں صرف ایک SNP پر ڈیٹا کی ضرورت ہوگی۔ مجھے یہ سیکھنا تھا کہ 2,5 ملین SNPs میں سے کسی ایک سے منسلک تمام ریکارڈز کو جتنی آسانی سے، جلدی اور سستے سے ممکن ہو سکے بازیافت کرنا ہے۔
یہ کیسے نہیں کرنا ہے۔
ایک مناسب کلچ کا حوالہ دینے کے لیے:
میں ایک ہزار بار ناکام نہیں ہوا، میں نے سوال کے موافق فارمیٹ میں ڈیٹا کے ایک گروپ کو پارس کرنے سے بچنے کے لیے صرف ایک ہزار طریقے دریافت کیے ہیں۔
پہلی کوشش
میں نے کیا سیکھا ہے۔: ایک وقت میں 25 ٹی بی کو پارس کرنے کا کوئی سستا طریقہ نہیں ہے۔
وینڈربلٹ یونیورسٹی میں "بگ ڈیٹا پروسیسنگ کے جدید طریقے" کورس کرنے کے بعد، مجھے یقین تھا کہ یہ چال تھیلے میں تھی۔ تمام ڈیٹا کو چلانے اور نتیجہ کی اطلاع دینے میں Hive سرور کو ترتیب دینے میں شاید ایک یا دو گھنٹے لگیں گے۔ چونکہ ہمارا ڈیٹا AWS S3 میں محفوظ ہے، اس لیے میں نے سروس استعمال کی۔ ، جو آپ کو S3 ڈیٹا پر Hive SQL سوالات لاگو کرنے کی اجازت دیتا ہے۔ آپ کو Hive کلسٹر قائم کرنے/اٹھانے کی ضرورت نہیں ہے، اور آپ صرف اس ڈیٹا کے لیے ادائیگی کرتے ہیں جس کی آپ تلاش کر رہے ہیں۔
ایتھینا کو اپنا ڈیٹا اور اس کی شکل دکھانے کے بعد، میں نے اس طرح کے سوالات کے ساتھ کچھ ٹیسٹ چلائے:
select * from intensityData limit 10;اور فوری طور پر اچھی طرح سے منظم نتائج حاصل کیے. تیار.
جب تک ہم نے اپنے کام میں ڈیٹا استعمال کرنے کی کوشش نہیں کی...
مجھے ماڈل کو جانچنے کے لیے تمام SNP معلومات نکالنے کو کہا گیا۔ میں نے استفسار کیا:
select * from intensityData
where snp = 'rs123456';اور انتظار کرنے لگا۔ آٹھ منٹ اور درخواست کردہ ڈیٹا کے 4 TB سے زیادہ کے بعد، مجھے نتیجہ موصول ہوا۔ ایتھینا ڈیٹا کے حجم کے حساب سے چارج کرتا ہے، $5 فی ٹیرا بائٹ۔ لہذا اس ایک درخواست پر $20 اور آٹھ منٹ انتظار کی لاگت آئی۔ تمام ڈیٹا پر ماڈل چلانے کے لیے ہمیں 38 سال انتظار کرنا پڑا اور ظاہر ہے کہ یہ ہمارے لیے مناسب نہیں تھا۔
Parquet استعمال کرنا ضروری تھا...
میں نے کیا سیکھا ہے۔: اپنی Parquet فائلوں کے سائز اور ان کی تنظیم کے بارے میں محتاط رہیں۔
میں نے سب سے پہلے تمام TSVs کو تبدیل کرکے صورتحال کو ٹھیک کرنے کی کوشش کی۔ . وہ بڑے ڈیٹا سیٹس کے ساتھ کام کرنے کے لیے آسان ہیں کیونکہ ان میں موجود معلومات کالمی شکل میں محفوظ ہوتی ہیں: ہر کالم اپنی میموری/ڈسک سیگمنٹ میں ہوتا ہے، ٹیکسٹ فائلوں کے برعکس، جس میں قطاروں میں ہر کالم کے عناصر ہوتے ہیں۔ اور اگر آپ کو کچھ تلاش کرنا ہو تو صرف مطلوبہ کالم پڑھیں۔ مزید برآں، ہر فائل ایک کالم میں قدروں کی ایک رینج کو اسٹور کرتی ہے، لہذا اگر آپ جس قدر کی تلاش کر رہے ہیں وہ کالم کی حد میں نہیں ہے، تو Spark پوری فائل کو اسکین کرنے میں وقت ضائع نہیں کرے گا۔
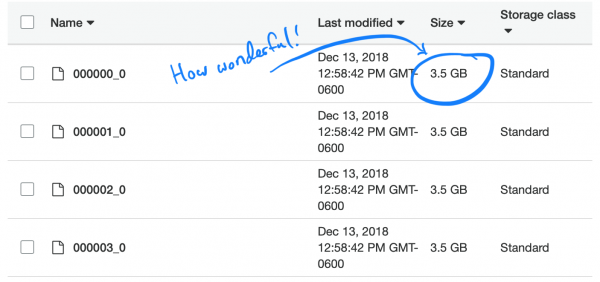
میں نے ایک آسان کام انجام دیا۔ ہماری TSVs کو Parquet میں تبدیل کرنے اور نئی فائلوں کو ایتھینا میں چھوڑنے کے لیے۔ اس میں تقریباً 5 گھنٹے لگے۔ لیکن جب میں نے درخواست چلائی، تو اسے مکمل کرنے میں تقریباً اتنا ہی وقت اور تھوڑا کم پیسہ لگا۔ حقیقت یہ ہے کہ اسپارک نے، کام کو بہتر بنانے کی کوشش کرتے ہوئے، صرف ایک TSV ٹکڑا کھولا اور اسے اپنے Parquet حصے میں ڈال دیا۔ اور چونکہ ہر ایک حصہ اتنا بڑا تھا کہ بہت سے لوگوں کے پورے ریکارڈ پر مشتمل ہو، ہر فائل میں تمام SNPs موجود تھے، اس لیے اسپارک کو اپنی مطلوبہ معلومات نکالنے کے لیے تمام فائلوں کو کھولنا پڑا۔
دلچسپ بات یہ ہے کہ پارکیٹ کی ڈیفالٹ (اور تجویز کردہ) کمپریشن کی قسم، تیز، تقسیم کے قابل نہیں ہے۔ لہذا، ہر ایگزیکیٹر مکمل 3,5 جی بی ڈیٹاسیٹ کو پیک کھولنے اور ڈاؤن لوڈ کرنے کے کام پر پھنس گیا تھا۔
آئیے مسئلہ کو سمجھتے ہیں۔
میں نے کیا سیکھا ہے۔: چھانٹنا مشکل ہے، خاص طور پر اگر ڈیٹا تقسیم کیا جائے۔
مجھے ایسا لگ رہا تھا کہ اب میں مسئلے کا نچوڑ سمجھ گیا ہوں۔ مجھے صرف SNP کالم کے ذریعہ ڈیٹا کو ترتیب دینے کی ضرورت تھی، لوگوں کے ذریعہ نہیں۔ اس کے بعد متعدد SNPs کو ایک علیحدہ ڈیٹا ٹکڑا میں ذخیرہ کیا جائے گا، اور پھر Parquet کا "سمارٹ" فنکشن "صرف اس صورت میں کھلتا ہے جب قیمت رینج میں ہو" اپنے آپ کو پوری شان و شوکت میں ظاہر کرے گا۔ بدقسمتی سے، ایک کلسٹر میں بکھری ہوئی اربوں قطاروں کو چھانٹنا ایک مشکل کام ثابت ہوا۔
میں کالج میں الگورتھم کی کلاس لے رہا ہوں: "اوہ، کسی کو ان تمام الگورتھم چھانٹنے کی کمپیوٹیشنل پیچیدگی کی پرواہ نہیں ہے"
میں 20TB میں کالم کو ترتیب دینے کی کوشش کر رہا ہوں۔ میز: "یہ اتنا وقت کیوں لے رہا ہے؟" جدوجہد.
— نک اسٹریر (@ نکولس اسٹریر)
AWS یقینی طور پر "میں ایک پریشان طالب علم ہوں" کی وجہ سے رقم کی واپسی جاری نہیں کرنا چاہتا۔ ایمیزون گلو پر چھانٹنے کے بعد، یہ 2 دن تک چلتا رہا اور کریش ہوگیا۔
تقسیم کے بارے میں کیا خیال ہے؟
میں نے کیا سیکھا ہے۔: سپارک میں پارٹیشنز کو متوازن ہونا چاہیے۔
تب مجھے کروموسوم میں ڈیٹا تقسیم کرنے کا خیال آیا۔ ان میں سے 23 ہیں (اور اگر آپ مائٹوکونڈریل ڈی این اے اور بغیر نقشے والے خطوں کو مدنظر رکھیں تو کئی اور)۔
یہ آپ کو ڈیٹا کو چھوٹے حصوں میں تقسیم کرنے کی اجازت دے گا۔ اگر آپ گلو اسکرپٹ میں اسپارک ایکسپورٹ فنکشن میں صرف ایک لائن شامل کرتے ہیں۔ partition_by = "chr"، پھر ڈیٹا کو بالٹیوں میں تقسیم کیا جانا چاہئے۔
جینوم متعدد ٹکڑوں پر مشتمل ہوتا ہے جسے کروموسوم کہتے ہیں۔
بدقسمتی سے، اس نے کام نہیں کیا۔ کروموسوم مختلف سائز کے ہوتے ہیں، جس کا مطلب ہے معلومات کی مختلف مقدار۔ اس کا مطلب ہے کہ جو کام Spark نے کارکنوں کو بھیجے وہ متوازن نہیں تھے اور آہستہ آہستہ مکمل ہوئے کیونکہ کچھ نوڈس جلد ختم ہو گئے تھے اور بیکار تھے۔ تاہم کام مکمل ہو گئے۔ لیکن ایک SNP کے لیے پوچھنے پر، عدم توازن دوبارہ مسائل کا باعث بنا۔ بڑے کروموسوم (یعنی جہاں ہم ڈیٹا حاصل کرنا چاہتے ہیں) پر SNPs کی پروسیسنگ کی لاگت میں صرف 10 کے فیکٹر کی کمی آئی ہے۔ بہت کچھ، لیکن کافی نہیں۔
اگر ہم اسے چھوٹے حصوں میں تقسیم کر دیں تو کیا ہوگا؟
میں نے کیا سیکھا ہے۔: کبھی بھی 2,5 ملین پارٹیشنز کرنے کی کوشش نہ کریں۔
میں نے سب سے باہر جانے کا فیصلہ کیا اور ہر SNP کو تقسیم کیا۔ اس نے اس بات کو یقینی بنایا کہ پارٹیشنز برابر سائز کے تھے۔ یہ ایک برا خیال تھا۔. میں نے گلو کا استعمال کیا اور ایک معصوم لائن شامل کی۔ partition_by = 'snp'. کام شروع ہوا اور اس پر عملدرآمد شروع کر دیا۔ ایک دن بعد میں نے چیک کیا اور دیکھا کہ S3 پر ابھی تک کچھ نہیں لکھا گیا، اس لیے میں نے کام ختم کردیا۔ ایسا لگتا ہے کہ گلو انٹرمیڈیٹ فائلیں S3 میں ایک پوشیدہ مقام پر لکھ رہا تھا، بہت سی فائلیں، شاید چند ملین۔ نتیجے کے طور پر، میری غلطی کی قیمت ایک ہزار ڈالر سے زیادہ تھی اور اس نے میرے سرپرست کو خوش نہیں کیا۔
تقسیم کرنا + چھانٹنا
میں نے کیا سیکھا ہے۔: چھانٹنا اب بھی مشکل ہے، جیسا کہ اسپارک کو ٹیوننگ کرنا ہے۔
تقسیم کرنے کی میری آخری کوشش میں کروموسوم کو تقسیم کرنا اور پھر ہر پارٹیشن کو ترتیب دینا شامل تھا۔ اصولی طور پر، یہ ہر استفسار کو تیز کر دے گا کیونکہ مطلوبہ SNP ڈیٹا کو ایک مخصوص حد کے اندر چند Parquet ٹکڑوں کے اندر ہونا چاہیے۔ بدقسمتی سے، تقسیم شدہ ڈیٹا کو بھی چھانٹنا ایک مشکل کام نکلا۔ نتیجے کے طور پر، میں نے حسب ضرورت کلسٹر کے لیے EMR پر سوئچ کیا اور مزید لچکدار ورک فلو بنانے کے لیے آٹھ طاقتور مثالیں (C5.4xl) اور Sparklyr کا استعمال کیا۔
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)تاہم، کام ابھی تک مکمل نہیں ہوا تھا۔ میں نے اسے مختلف طریقوں سے ترتیب دیا: ہر استفسار پر عمل کرنے والے کے لیے میموری مختص میں اضافہ کیا، بڑی مقدار میں میموری کے ساتھ نوڈس کا استعمال کیا، براڈکاسٹ متغیرات (براڈکاسٹنگ متغیرات) کا استعمال کیا، لیکن ہر بار یہ آدھے اقدامات کے طور پر نکلے، اور دھیرے دھیرے ایگزیکیوٹرز شروع ہوئے۔ ناکام ہونے کے لئے جب تک سب کچھ بند نہ ہو.
اپ ڈیٹ: تو یہ شروع ہوتا ہے۔
— نک اسٹریر (@ نکولس اسٹریر)
میں زیادہ تخلیقی ہوتا جا رہا ہوں۔
میں نے کیا سیکھا ہے۔: بعض اوقات خصوصی ڈیٹا کے لیے خصوصی حل کی ضرورت ہوتی ہے۔
ہر SNP کی پوزیشن ویلیو ہوتی ہے۔ یہ ایک عدد ہے جو اس کے کروموسوم کے ساتھ اڈوں کی تعداد کے مطابق ہے۔ یہ ہمارے ڈیٹا کو منظم کرنے کا ایک اچھا اور قدرتی طریقہ ہے۔ پہلے میں ہر کروموسوم کے علاقوں کے لحاظ سے تقسیم کرنا چاہتا تھا۔ مثال کے طور پر، پوزیشنز 1 - 2000، 2001 - 4000، وغیرہ۔ لیکن مسئلہ یہ ہے کہ SNPs کو کروموسوم میں یکساں طور پر تقسیم نہیں کیا جاتا ہے، اس لیے گروپ کے سائز بہت مختلف ہوں گے۔
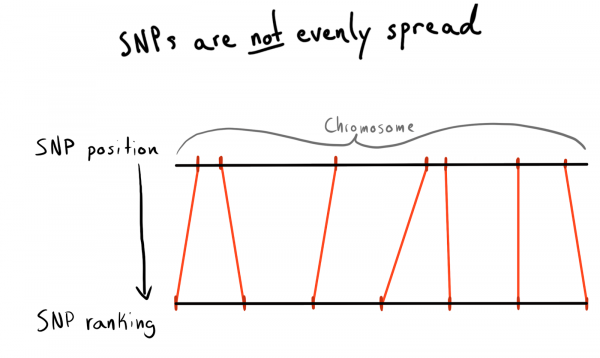
نتیجے کے طور پر، میں عہدوں کو زمرے (درجہ) میں تقسیم کر کے آیا۔ پہلے سے ڈاؤن لوڈ کردہ ڈیٹا کا استعمال کرتے ہوئے، میں نے منفرد SNPs، ان کی پوزیشنوں اور کروموسوم کی فہرست حاصل کرنے کی درخواست کی۔ پھر میں نے ہر کروموسوم کے اندر ڈیٹا کو ترتیب دیا اور SNPs کو ایک مخصوص سائز کے گروپس (بن) میں جمع کیا۔ آئیے ہر ایک کو 1000 SNPs کہتے ہیں۔ اس نے مجھے SNP سے گروپ فی کروموسوم رشتہ دیا۔
آخر میں، میں نے 75 SNPs کے گروپ (بن) بنائے، اس کی وجہ ذیل میں بیان کی جائے گی۔
snp_to_bin <- unique_snps %>%
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()پہلے Spark کے ساتھ کوشش کریں۔
میں نے کیا سیکھا ہے۔: چنگاری جمع کرنا تیز ہے، لیکن تقسیم اب بھی مہنگا ہے۔
میں اس چھوٹے (2,5 ملین قطاروں) کے ڈیٹا فریم کو سپارک میں پڑھنا چاہتا تھا، اسے خام ڈیٹا کے ساتھ جوڑنا چاہتا تھا، اور پھر اسے نئے شامل کیے گئے کالم کے ذریعے تقسیم کرنا چاہتا تھا۔ bin.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
)
میں نے استعمال کیا sdf_broadcast()، لہذا اسپارک جانتا ہے کہ اسے تمام نوڈس کو ڈیٹا فریم بھیجنا چاہئے۔ یہ مفید ہے اگر ڈیٹا سائز میں چھوٹا ہو اور تمام کاموں کے لیے درکار ہو۔ بصورت دیگر، اسپارک اسمارٹ بننے کی کوشش کرتا ہے اور ضرورت کے مطابق ڈیٹا تقسیم کرتا ہے، جو سست روی کا سبب بن سکتا ہے۔
اور ایک بار پھر، میرا خیال کام نہیں کر سکا: کاموں نے کچھ عرصے تک کام کیا، یونین کو مکمل کیا، اور پھر، تقسیم کے ذریعے شروع کیے گئے عملداروں کی طرح، وہ ناکام ہونے لگے۔
AWK شامل کرنا
میں نے کیا سیکھا ہے۔: جب آپ کو بنیادی باتیں سکھائی جائیں تو نہ سوئے۔ یقیناً کسی نے آپ کا مسئلہ 1980 کی دہائی میں حل کر دیا ہے۔
اس وقت تک، اسپارک کے ساتھ میری تمام ناکامیوں کی وجہ کلسٹر میں ڈیٹا کا گڑبڑ تھا۔ شاید پہلے سے علاج سے صورتحال کو بہتر کیا جا سکتا ہے۔ میں نے خام ٹیکسٹ ڈیٹا کو کروموسوم کے کالموں میں تقسیم کرنے کی کوشش کرنے کا فیصلہ کیا، اس لیے میں نے اسپارک کو "پہلے سے تقسیم شدہ" ڈیٹا فراہم کرنے کی امید ظاہر کی۔
میں نے اسٹیک اوور فلو پر تلاش کیا کہ کالم کی قدروں سے کیسے تقسیم کیا جائے اور ملا AWK کے ساتھ آپ ٹیکسٹ فائل کو کالم کی قدروں کے مطابق اسکرپٹ میں لکھ کر تقسیم کر سکتے ہیں بجائے اس کے کہ نتائج بھیجیں۔ stdout.
میں نے اسے آزمانے کے لیے باش اسکرپٹ لکھا۔ پیکیج شدہ TSVs میں سے ایک کو ڈاؤن لوڈ کیا، پھر اسے استعمال کرکے کھولیں۔ gzip اور بھیج دیا awk.
gzip -dc path/to/chunk/file.gz |
awk -F 't'
'{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'یہ کام کر گیا!
کور کو بھرنا
میں نے کیا سیکھا ہے۔: gnu parallel - یہ ایک جادوئی چیز ہے، ہر ایک کو اسے استعمال کرنا چاہیے۔
علیحدگی کافی سست تھی اور جب میں نے شروع کیا۔ htopایک طاقتور (اور مہنگی) EC2 مثال کے استعمال کو چیک کرنے کے لیے، پتہ چلا کہ میں صرف ایک کور اور تقریباً 200 MB میموری استعمال کر رہا ہوں۔ مسئلہ کو حل کرنے اور بہت سارے پیسے کھونے کے لئے، ہمیں کام کو متوازی کرنے کا طریقہ معلوم کرنا پڑا۔ خوش قسمتی سے، ایک بالکل حیرت انگیز کتاب میں مجھے متوازی پر جیرون جانسنز کا ایک باب ملا۔ اس سے میں نے سیکھا۔ gnu parallelیونکس میں ملٹی تھریڈنگ کو لاگو کرنے کا ایک بہت ہی لچکدار طریقہ۔

جب میں نے نئے عمل کا استعمال کرتے ہوئے پارٹیشننگ شروع کی تو سب کچھ ٹھیک تھا، لیکن پھر بھی ایک رکاوٹ تھی - S3 اشیاء کو ڈسک پر ڈاؤن لوڈ کرنا بہت تیز نہیں تھا اور مکمل طور پر متوازی نہیں تھا۔ اسے ٹھیک کرنے کے لیے، میں نے یہ کیا:
- مجھے پتہ چلا کہ S3 ڈاؤن لوڈ کے مرحلے کو براہ راست پائپ لائن میں لاگو کرنا ممکن ہے، ڈسک پر انٹرمیڈیٹ اسٹوریج کو مکمل طور پر ختم کر کے۔ اس کا مطلب ہے کہ میں ڈسک پر خام ڈیٹا لکھنے سے بچ سکتا ہوں اور اس سے بھی چھوٹا، اور اس وجہ سے سستا، AWS پر اسٹوریج استعمال کر سکتا ہوں۔
- ٹیم
aws configure set default.s3.max_concurrent_requests 50AWS CLI استعمال کرنے والے تھریڈز کی تعداد میں بہت زیادہ اضافہ کر دیا ہے (بطور ڈیفالٹ 10 ہیں)۔ - میں نے نام میں n حرف کے ساتھ، نیٹ ورک کی رفتار کے لیے موزوں کردہ EC2 مثال پر سوئچ کیا۔ میں نے محسوس کیا ہے کہ n-انسٹینس استعمال کرتے وقت پروسیسنگ پاور کا نقصان لوڈنگ کی رفتار میں اضافے سے ہونے والی تلافی سے زیادہ ہے۔ زیادہ تر کاموں کے لیے میں نے c5n.4xl استعمال کیا۔
- بدل گیا۔
gzipپر ، یہ ایک gzip ٹول ہے جو فائلوں کو ڈیکمپریس کرنے کے ابتدائی طور پر غیر متوازی کام کو متوازی کرنے کے لئے ٹھنڈی چیزیں کرسکتا ہے (اس سے کم سے کم مدد ہوئی)۔
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe
"awk -F 't' '{print $1",..."$30">"chunked/{#}_chr"$15".csv"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneہر چیز کو بہت تیزی سے کام کرنے کے لیے یہ اقدامات ایک دوسرے کے ساتھ مل جاتے ہیں۔ ڈاؤن لوڈ کی رفتار میں اضافہ کرکے اور ڈسک رائٹ کو ختم کرکے، میں اب صرف چند گھنٹوں میں 5 ٹیرا بائٹ پیکج پر کارروائی کرسکتا ہوں۔
AWS پر استعمال ہونے والے تمام کور کو دیکھنے سے زیادہ میٹھی چیز نہیں ہے۔ gnu-paralel کی بدولت میں 19gig csv کو ان زپ اور تقسیم کر سکتا ہوں جتنی تیزی سے میں اسے ڈاؤن لوڈ کر سکتا ہوں۔ مجھے اس کو چلانے کے لیے چنگاری بھی نہیں مل سکی۔
— نک اسٹریر (@ نکولس اسٹریر)
اس ٹویٹ میں 'TSV' کا ذکر ہونا چاہیے تھا۔ افسوس
نئے تجزیہ کردہ ڈیٹا کا استعمال
میں نے کیا سیکھا ہے۔: اسپارک غیر کمپریسڈ ڈیٹا کو پسند کرتا ہے اور پارٹیشنز کو یکجا کرنا پسند نہیں کرتا۔
اب ڈیٹا S3 میں غیر پیک شدہ (پڑھیں: مشترکہ) اور نیم ترتیب شدہ فارمیٹ میں تھا، اور میں دوبارہ سپارک پر واپس جا سکتا تھا۔ ایک حیرت میرا انتظار کر رہی تھی: میں ایک بار پھر وہ حاصل کرنے میں ناکام رہا جو میں چاہتا تھا! اسپارک کو یہ بتانا بہت مشکل تھا کہ ڈیٹا کو کیسے تقسیم کیا گیا تھا۔ اور جب میں نے یہ کیا تو پتہ چلا کہ پارٹیشنز بہت زیادہ ہیں (95 ہزار) اور جب میں نے استعمال کیا coalesce ان کی تعداد کو مناسب حد تک کم کر دیا، اس نے میری تقسیم کو تباہ کر دیا۔ مجھے یقین ہے کہ یہ ٹھیک ہو سکتا ہے، لیکن چند دنوں کی تلاش کے بعد بھی مجھے کوئی حل نہیں مل سکا۔ میں نے بالآخر اسپارک میں تمام کام مکمل کر لیے، حالانکہ اس میں کچھ وقت لگا اور میری اسپلٹ پارکیٹ فائلیں بہت چھوٹی نہیں تھیں (~200 KB)۔ تاہم، ڈیٹا وہیں تھا جہاں اس کی ضرورت تھی۔
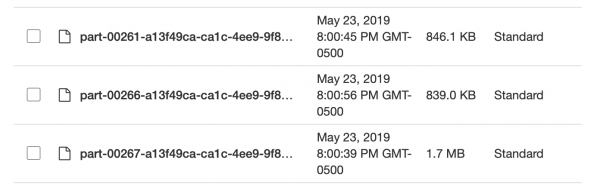
بہت چھوٹا اور ناہموار، شاندار!
مقامی اسپارک سوالات کی جانچ کرنا
میں نے کیا سیکھا ہے۔: سادہ مسائل کو حل کرتے وقت چنگاری کا سر بہت زیادہ ہوتا ہے۔
ہوشیار فارمیٹ میں ڈیٹا ڈاؤن لوڈ کرکے، میں رفتار کو جانچنے کے قابل تھا۔ مقامی اسپارک سرور کو چلانے کے لیے ایک R اسکرپٹ مرتب کریں، اور پھر مخصوص پارکیٹ گروپ اسٹوریج (بن) سے اسپارک ڈیٹا فریم لوڈ کریں۔ میں نے تمام ڈیٹا لوڈ کرنے کی کوشش کی لیکن اسپارکلیر کو تقسیم کو پہچاننے کے لیے نہیں مل سکا۔
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Start a timer
start_time <- Sys.time()
# Load the desired bin into Spark
intensity_data <- sc %>%
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Subset bin to snp and then collect to local
test_subset <- intensity_data %>%
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)پھانسی میں 29,415 سیکنڈ لگے۔ بہت بہتر، لیکن کسی بھی چیز کی بڑے پیمانے پر جانچ کے لیے بہت اچھا نہیں۔ مزید برآں، میں کیشنگ کے ساتھ چیزوں کو تیز نہیں کر سکا کیونکہ جب میں نے میموری میں ڈیٹا فریم کو کیش کرنے کی کوشش کی، تو Spark ہمیشہ کریش ہو جاتا ہے، یہاں تک کہ جب میں نے 50 GB سے زیادہ میموری کو ڈیٹا سیٹ کے لیے مختص کیا جس کا وزن 15 سے کم تھا۔
AWK پر واپس جائیں۔
میں نے کیا سیکھا ہے۔: AWK میں ایسوسی ایٹیو صفیں بہت موثر ہیں۔
میں نے محسوس کیا کہ میں تیز رفتاری حاصل کرسکتا ہوں۔ میں نے ایک شاندار میں یاد کیا میں نے ایک عمدہ خصوصیت کے بارے میں پڑھا جسے "" بنیادی طور پر، یہ کلیدی قدر کے جوڑے ہیں، جنہیں کسی وجہ سے AWK میں مختلف کہا جاتا تھا، اور اس وجہ سے میں نے ان کے بارے میں زیادہ نہیں سوچا۔ یاد آیا کہ "ایسوسی ایٹیو اریز" کی اصطلاح "کلیدی قدر کی جوڑی" کی اصطلاح سے بہت پرانی ہے۔ یہاں تک کہ اگر آپ ، آپ کو یہ اصطلاح وہاں نظر نہیں آئے گی، لیکن آپ کو ایسوسی ایٹیو صفیں ملیں گی! اس کے علاوہ، "کلیدی قدر کی جوڑی" اکثر ڈیٹا بیس کے ساتھ منسلک ہوتی ہے، لہذا اس کا موازنہ ہیش میپ سے کرنا بہت زیادہ معنی خیز ہے۔ میں نے محسوس کیا کہ میں Spark کو استعمال کیے بغیر اپنے SNPs کو بِن ٹیبل اور خام ڈیٹا کے ساتھ منسلک کرنے کے لیے ان ایسوسی ایٹیو صفوں کا استعمال کر سکتا ہوں۔
ایسا کرنے کے لیے، AWK اسکرپٹ میں میں نے بلاک استعمال کیا۔ BEGIN. یہ کوڈ کا ایک ٹکڑا ہے جو اسکرپٹ کے مین باڈی میں ڈیٹا کی پہلی لائن کو منتقل کرنے سے پہلے عمل میں لایا جاتا ہے۔
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline < "snp_to_bin.csv") {bin[$1] = $2}
}
{
print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
}
ٹیم while(getline...) CSV گروپ (بن) سے تمام قطاریں لوڈ کیں، پہلے کالم (SNP نام) کو ایسوسی ایٹیو صف کے لیے کلید کے طور پر سیٹ کریں۔ bin اور دوسری قدر (گروپ) بطور قدر۔ پھر بلاک میں { }، جو مین فائل کی تمام لائنوں پر عمل میں لایا جاتا ہے، ہر لائن آؤٹ پٹ فائل کو بھیجی جاتی ہے، جو اپنے گروپ (بن) کے لحاظ سے ایک منفرد نام حاصل کرتی ہے۔ ..._bin_"bin[$1]"_....
متغیرات۔ batch_num и chunk_id پائپ لائن کے ذریعہ فراہم کردہ ڈیٹا سے مماثل ہے، ریس کی حالت سے گریز کرتے ہوئے، اور ہر ایک ایگزیکیوشن تھریڈ چل رہا ہے۔ parallel، نے اپنی منفرد فائل میں لکھا۔
چونکہ میں نے AWK کے ساتھ اپنے پچھلے تجربے سے رہ جانے والے تمام خام ڈیٹا کو کروموسوم پر فولڈرز میں بکھیر دیا تھا، اب میں ایک وقت میں ایک کروموسوم پر کارروائی کرنے کے لیے ایک اور باش اسکرپٹ لکھ سکتا ہوں اور گہرا تقسیم شدہ ڈیٹا S3 کو بھیج سکتا ہوں۔
DESIRED_CHR='13'
# Download chromosome data from s3 and split into bins
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'reading {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=""$DESIRED_CHR"" -v chunk="{}" -f split_on_chr_bin.awk"
# Combine all the parallel process chunks to single files and upload to rds using R
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/*
اسکرپٹ کے دو حصے ہیں۔ parallel.
پہلے حصے میں، مطلوبہ کروموسوم کی معلومات پر مشتمل تمام فائلوں سے ڈیٹا پڑھا جاتا ہے، پھر اس ڈیٹا کو تھریڈز میں تقسیم کیا جاتا ہے، جو فائلوں کو مناسب گروپس (بن) میں تقسیم کرتے ہیں۔ ریس کے حالات سے بچنے کے لیے جب ایک سے زیادہ تھریڈز ایک ہی فائل پر لکھتے ہیں، AWK مختلف جگہوں پر ڈیٹا لکھنے کے لیے فائل کے ناموں کو پاس کرتا ہے، جیسے chr_10_bin_52_batch_2_aa.csv. نتیجے کے طور پر، بہت سی چھوٹی فائلیں ڈسک پر بنتی ہیں (اس کے لیے میں نے ٹیرابائٹ ای بی ایس والیوم استعمال کیے)۔
دوسرے حصے سے کنویئر parallel گروپس (بن) سے گزرتا ہے اور ان کی انفرادی فائلوں کو مشترکہ CSV c میں جوڑتا ہے۔ catاور پھر برآمد کے لیے بھیجتا ہے۔
آر میں نشریات؟
میں نے کیا سیکھا ہے۔: آپ رابطہ کر سکتے ہیں۔ stdin и stdout ایک R اسکرپٹ سے، اور اس لیے اسے پائپ لائن میں استعمال کریں۔
آپ نے اپنے باش اسکرپٹ میں اس لائن کو دیکھا ہوگا۔ ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... یہ تمام مربوط گروپ فائلوں (بن) کا ذیل میں R اسکرپٹ میں ترجمہ کرتا ہے۔ {} ایک خاص تکنیک ہے parallel، جو کسی بھی ڈیٹا کو داخل کرتا ہے جسے وہ مخصوص اسٹریم کو بھیجتا ہے براہ راست کمانڈ میں ہی۔ آپشن {#} ایک منفرد تھریڈ ID فراہم کرتا ہے، اور {%} جاب سلاٹ نمبر کی نمائندگی کرتا ہے (بار بار، لیکن بیک وقت کبھی نہیں)۔ تمام اختیارات کی فہرست میں مل سکتی ہے۔
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
)
جب ایک متغیر file("stdin") کو منتقل کیا گیا ہے۔ readr::read_csv، R اسکرپٹ میں ترجمہ کردہ ڈیٹا کو ایک فریم میں لوڈ کیا جاتا ہے، جو پھر فارم میں ہوتا ہے۔ .rdsفائل کا استعمال کرتے ہوئے aws.s3 براہ راست S3 پر لکھا گیا۔
RDS Parquet کے جونیئر ورژن کی طرح کچھ ہے، بغیر اسپیکر اسٹوریج کے۔
باش اسکرپٹ کو ختم کرنے کے بعد مجھے ایک بنڈل ملا .rdsS3 میں واقع فائلیں، جس نے مجھے موثر کمپریشن اور بلٹ ان اقسام استعمال کرنے کی اجازت دی۔
بریک R کے استعمال کے باوجود، سب کچھ بہت تیزی سے کام کرتا ہے۔ حیرت کی بات نہیں ہے، R کے وہ حصے جو ڈیٹا کو پڑھتے اور لکھتے ہیں بہت زیادہ بہتر ہوتے ہیں۔ ایک درمیانے سائز کے کروموسوم پر ٹیسٹ کرنے کے بعد، کام تقریباً دو گھنٹے میں C5n.4xl مثال پر مکمل ہو گیا۔
S3 حدود
میں نے کیا سیکھا ہے۔: سمارٹ پاتھ کے نفاذ کی بدولت، S3 بہت سی فائلوں کو سنبھال سکتا ہے۔
میں پریشان تھا کہ آیا S3 ان بہت سی فائلوں کو سنبھال سکے گا جو اس میں منتقل کی گئی تھیں۔ میں فائل کے ناموں کو معنی خیز بنا سکتا ہوں، لیکن S3 انہیں کیسے تلاش کرے گا؟

S3 میں فولڈرز صرف دکھانے کے لیے ہیں، درحقیقت سسٹم کو علامت میں کوئی دلچسپی نہیں ہے۔ /.
ایسا لگتا ہے کہ S3 ہیش ٹیبل یا دستاویز پر مبنی ڈیٹا بیس میں ایک سادہ کلید کے طور پر کسی مخصوص فائل کے راستے کی نمائندگی کرتا ہے۔ ایک بالٹی کو ایک میز کے طور پر سمجھا جا سکتا ہے، اور فائلوں کو اس ٹیبل میں ریکارڈ سمجھا جا سکتا ہے۔
چونکہ ایمیزون پر منافع کمانے کے لیے رفتار اور کارکردگی اہم ہے، اس لیے اس میں کوئی تعجب کی بات نہیں ہے کہ یہ کلید کے طور پر فائل پاتھ سسٹم کو بہتر بنایا جا رہا ہے۔ میں نے بیلنس تلاش کرنے کی کوشش کی: تاکہ مجھے بہت زیادہ درخواستیں نہیں کرنی پڑیں، لیکن یہ کہ درخواستوں پر تیزی سے عمل درآمد ہو گیا۔ معلوم ہوا کہ تقریباً 20 ہزار بن فائلیں بنانا بہترین ہے۔ میرا خیال ہے کہ اگر ہم اصلاح کرنا جاری رکھیں تو ہم رفتار میں اضافہ حاصل کر سکتے ہیں (مثال کے طور پر، صرف ڈیٹا کے لیے ایک خاص بالٹی بنانا، اس طرح تلاش کی میز کے سائز کو کم کرنا)۔ لیکن مزید تجربات کے لیے وقت یا پیسہ نہیں تھا۔
کراس مطابقت کے بارے میں کیا خیال ہے؟
میں نے کیا سیکھا: وقت کے ضائع ہونے کی پہلی وجہ وقت سے پہلے آپ کے اسٹوریج کے طریقہ کار کو بہتر بنانا ہے۔
اس موقع پر، اپنے آپ سے یہ پوچھنا بہت ضروری ہے: "ایک ملکیتی فائل فارمیٹ کیوں استعمال کریں؟" اس کی وجہ لوڈنگ کی رفتار (gzipped CSV فائلوں کو لوڈ ہونے میں 7 گنا زیادہ وقت لگا) اور ہمارے ورک فلو کے ساتھ مطابقت ہے۔ میں اس پر دوبارہ غور کر سکتا ہوں کہ آیا R Spark لوڈ کے بغیر Parquet (یا Arrow) فائلوں کو آسانی سے لوڈ کر سکتا ہے۔ ہماری لیب میں ہر کوئی R کا استعمال کرتا ہے، اور اگر مجھے ڈیٹا کو کسی اور فارمیٹ میں تبدیل کرنے کی ضرورت ہے، تو میرے پاس اب بھی اصل ٹیکسٹ ڈیٹا موجود ہے، لہذا میں دوبارہ پائپ لائن چلا سکتا ہوں۔
کام کی تقسیم
میں نے کیا سیکھا ہے۔: ملازمتوں کو دستی طور پر بہتر بنانے کی کوشش نہ کریں، کمپیوٹر کو کرنے دیں۔
میں نے ایک کروموسوم پر ورک فلو کو ڈیبگ کیا ہے، اب مجھے باقی تمام ڈیٹا پر کارروائی کرنے کی ضرورت ہے۔
میں تبادلوں کے لیے کئی EC2 مثالیں اٹھانا چاہتا تھا، لیکن اس کے ساتھ ہی مجھے مختلف پروسیسنگ جابز میں بہت زیادہ غیر متوازن بوجھ پڑنے کا ڈر تھا (جس طرح اسپارک کو غیر متوازن پارٹیشنز کا سامنا کرنا پڑا تھا)۔ اس کے علاوہ، میں فی کروموسوم ایک مثال بڑھانے میں دلچسپی نہیں رکھتا تھا، کیونکہ AWS اکاؤنٹس کے لیے 10 مثالوں کی ڈیفالٹ حد ہوتی ہے۔
پھر میں نے پروسیسنگ جابز کو بہتر بنانے کے لیے R میں اسکرپٹ لکھنے کا فیصلہ کیا۔
سب سے پہلے، میں نے S3 سے حساب لگانے کے لیے کہا کہ ہر کروموسوم نے کتنی اسٹوریج کی جگہ لی ہے۔
library(aws.s3)
library(tidyverse)
chr_sizes <- get_bucket_df(
bucket = '...', prefix = '...', max = Inf
) %>%
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Extract chromosome from the file name
chr = str_extract(Key, 'chr.{1,4}.csv') %>%
str_remove_all('chr|.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Divide to get value in GB
# A tibble: 27 x 2
chr total_size
<chr> <dbl>
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … with 17 more rows
پھر میں نے ایک فنکشن لکھا جو کل سائز لیتا ہے، کروموسوم کی ترتیب کو بدلتا ہے، انہیں گروپوں میں تقسیم کرتا ہے۔ num_jobs اور آپ کو بتاتا ہے کہ تمام پروسیسنگ جابز کے سائز کتنے مختلف ہیں۔
num_jobs <- 7
# How big would each job be if perfectly split?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job <- function(i){
chr_sizes %>%
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)
# A tibble: 1 x 2
sd data
<dbl> <list>
1 153. <tibble [7 × 3]>پھر میں نے purrr کا استعمال کرتے ہوئے ایک ہزار شفلز کے ذریعے بھاگ کر بہترین کا انتخاب کیا۔
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1)
لہذا میں نے کاموں کے ایک سیٹ کے ساتھ ختم کیا جو سائز میں بہت ملتے جلتے تھے۔ پھر جو کچھ بچا تھا وہ یہ تھا کہ میری پچھلی باش اسکرپٹ کو ایک بڑے لوپ میں لپیٹنا تھا۔ for. اس اصلاح کو لکھنے میں تقریباً 10 منٹ لگے۔ اور یہ اس سے بہت کم ہے جو میں دستی طور پر کاموں کو بنانے پر خرچ کروں گا اگر وہ غیر متوازن ہوں۔ لہذا، میں سمجھتا ہوں کہ میں اس ابتدائی اصلاح کے ساتھ صحیح تھا۔
for DESIRED_CHR in "16" "9" "7" "21" "MT"
do
# Code for processing a single chromosome
fiآخر میں میں شٹ ڈاؤن کمانڈ شامل کرتا ہوں:
sudo shutdown -h now
... اور سب کچھ کام کیا! AWS CLI کا استعمال کرتے ہوئے، میں نے آپشن کا استعمال کرتے ہوئے مثالیں اٹھائیں user_data پروسیسنگ کے لیے انہیں ان کے کاموں کی Bash اسکرپٹس دیں۔ وہ بھاگ گئے اور خود بخود بند ہو گئے، اس لیے میں اضافی پروسیسنگ پاور کے لیے ادائیگی نہیں کر رہا تھا۔
aws ec2 run-instances ...
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]"
--user-data file://<<job_script_loc>>آئیے پیک کریں!
میں نے کیا سیکھا ہے۔: API کو استعمال میں آسانی اور لچک کی خاطر سادہ ہونا چاہیے۔
آخر کار مجھے صحیح جگہ اور فارم میں ڈیٹا مل گیا۔ بس باقی رہ گیا ڈیٹا کو زیادہ سے زیادہ استعمال کرنے کے عمل کو آسان بنانا تھا تاکہ میرے ساتھیوں کے لیے آسانی ہو۔ میں درخواستیں بنانے کے لیے ایک سادہ API بنانا چاہتا تھا۔ اگر مستقبل میں میں اس سے سوئچ کرنے کا فیصلہ کرتا ہوں۔ .rds Parquet فائلوں کے لیے، پھر یہ میرے لیے مسئلہ ہونا چاہیے، میرے ساتھیوں کے لیے نہیں۔ اس کے لیے میں نے ایک اندرونی R پیکج بنانے کا فیصلہ کیا۔
ایک بہت ہی آسان پیکیج بنائیں اور دستاویز کریں جس میں کسی فنکشن کے ارد گرد منظم کردہ صرف چند ڈیٹا تک رسائی کے فنکشن ہوں۔ get_snp. میں نے اپنے ساتھیوں کے لیے ایک ویب سائٹ بھی بنائی ، تاکہ وہ آسانی سے مثالیں اور دستاویزات دیکھ سکیں۔
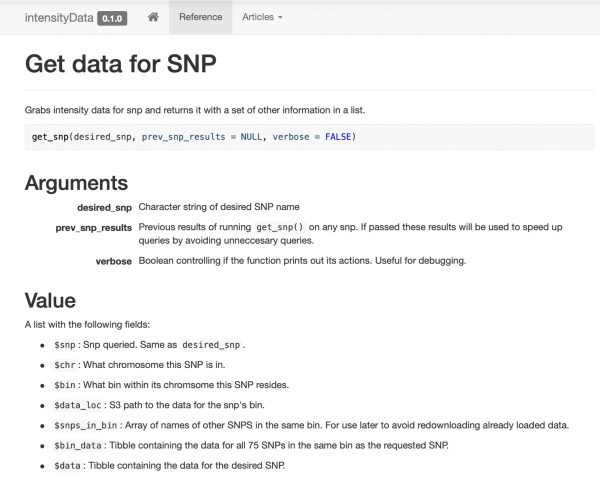
اسمارٹ کیشنگ
میں نے کیا سیکھا ہے۔: اگر آپ کا ڈیٹا اچھی طرح سے تیار ہے، تو کیشنگ آسان ہو جائے گی!
چونکہ ایک اہم ورک فلو نے اسی تجزیہ ماڈل کو SNP پیکیج پر لاگو کیا ہے، اس لیے میں نے اپنے فائدے کے لیے بائننگ استعمال کرنے کا فیصلہ کیا۔ SNP کے ذریعے ڈیٹا منتقل کرتے وقت، گروپ (بن) کی تمام معلومات واپس کی گئی آبجیکٹ کے ساتھ منسلک ہوتی ہیں۔ یعنی، پرانے سوالات (نظریہ میں) نئے سوالات کی کارروائی کو تیز کر سکتے ہیں۔
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
...
پیکج بناتے وقت، میں نے مختلف طریقوں کا استعمال کرتے ہوئے رفتار کا موازنہ کرنے کے لیے بہت سے بینچ مارکس چلائے۔ میں اس کو نظر انداز نہ کرنے کی سفارش کرتا ہوں، کیونکہ کبھی کبھی نتائج غیر متوقع ہیں. مثال کے طور پر، dplyr::filter اشاریہ سازی پر مبنی فلٹرنگ کا استعمال کرتے ہوئے قطاروں کو کیپچر کرنے سے کہیں زیادہ تیز تھا، اور فلٹر شدہ ڈیٹا فریم سے ایک کالم کو بازیافت کرنا اشاریہ سازی کی ترکیب استعمال کرنے سے کہیں زیادہ تیز تھا۔
براہ مہربانی نوٹ کریں کہ اعتراض prev_snp_results کلید پر مشتمل ہے snps_in_bin. یہ ایک گروپ (بن) میں تمام منفرد SNPs کی ایک صف ہے، جو آپ کو جلدی سے چیک کرنے کی اجازت دیتی ہے کہ آیا آپ کے پاس پہلے سے کسی سابقہ استفسار کا ڈیٹا موجود ہے۔ اس کوڈ کے ساتھ گروپ (بن) میں موجود تمام SNPs کو لوپ کرنا بھی آسان بناتا ہے۔
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}نتائج
اب ہم ایسے ماڈلز اور منظرنامے چلا سکتے ہیں (اور سنجیدگی سے شروع کر چکے ہیں) جو پہلے ہمارے لیے ناقابل رسائی تھے۔ سب سے اچھی بات یہ ہے کہ میرے لیب کے ساتھیوں کو کسی پیچیدگی کے بارے میں سوچنے کی ضرورت نہیں ہے۔ ان کے پاس صرف ایک فنکشن ہے جو کام کرتا ہے۔
اور اگرچہ پیکج ان کو تفصیلات سے بچاتا ہے، میں نے ڈیٹا فارمیٹ کو اتنا آسان بنانے کی کوشش کی کہ وہ اس بات کا اندازہ لگا سکیں کہ اگر میں کل اچانک غائب ہو گیا ہوں...
رفتار میں نمایاں اضافہ ہوا ہے۔ ہم عام طور پر فعال طور پر اہم جینوم کے ٹکڑوں کو اسکین کرتے ہیں۔ پہلے، ہم یہ نہیں کر سکتے تھے (یہ بہت مہنگا نکلا)، لیکن اب، گروپ (بن) کے ڈھانچے اور کیشنگ کی بدولت، ایک SNP کی درخواست میں اوسطاً 0,1 سیکنڈ سے بھی کم وقت لگتا ہے، اور ڈیٹا کا استعمال اتنا ہی ہے۔ کم ہے کہ S3 کی قیمت مونگ پھلی ہے۔
حال ہی میں میں نے اپنی لیب کے لیے 25+ TB خام جین ٹائپنگ ڈیٹا میں تبدیلی کی ہے۔ جب میں نے شروع کیا تو اسپارک کے استعمال میں 8 منٹ لگے اور SNP سے استفسار کرنے میں $20 لاگت آئی۔ AWK+ استعمال کرنے کے بعد اس پر کارروائی کرنے میں اب ایک سیکنڈ کے 10ویں حصے سے بھی کم وقت لگتا ہے اور اس کی قیمت $0.00001 ہے۔ میرا ذاتی جیت.
— نک اسٹریر (@ نکولس اسٹریر)
حاصل يہ ہوا
یہ مضمون بالکل بھی رہنما نہیں ہے۔ حل انفرادی طور پر نکلا، اور تقریبا یقینی طور پر زیادہ سے زیادہ نہیں ہے. بلکہ یہ ایک سفرنامہ ہے۔ میں چاہتا ہوں کہ دوسرے یہ سمجھیں کہ اس طرح کے فیصلے مکمل طور پر سر میں بنتے نظر نہیں آتے، یہ آزمائش اور غلطی کا نتیجہ ہوتے ہیں۔ اس کے علاوہ، اگر آپ ڈیٹا سائنسدان کی تلاش کر رہے ہیں، تو ذہن میں رکھیں کہ ان ٹولز کو مؤثر طریقے سے استعمال کرنے کے لیے تجربے کی ضرورت ہوتی ہے، اور تجربے کے لیے پیسے خرچ ہوتے ہیں۔ میں خوش ہوں کہ میرے پاس ادائیگی کرنے کا ذریعہ تھا، لیکن بہت سے دوسرے لوگ جو مجھ سے بہتر کام کر سکتے ہیں، پیسے کی کمی کی وجہ سے کبھی بھی کوشش کرنے کا موقع نہیں ملے گا۔
بڑے ڈیٹا ٹولز ورسٹائل ہیں۔ اگر آپ کے پاس وقت ہے، تو آپ یقینی طور پر اسمارٹ ڈیٹا کی صفائی، اسٹوریج اور نکالنے کی تکنیکوں کا استعمال کرتے ہوئے ایک تیز تر حل لکھ سکتے ہیں۔ بالآخر یہ لاگت سے فائدہ کے تجزیہ پر آتا ہے۔
میں نے کیا سیکھا:
- ایک وقت میں 25 ٹی بی کو پارس کرنے کا کوئی سستا طریقہ نہیں ہے۔
- اپنی پارکیٹ فائلوں کے سائز اور ان کی تنظیم سے محتاط رہیں۔
- اسپارک میں پارٹیشنز کو متوازن ہونا چاہیے۔
- عام طور پر، کبھی بھی 2,5 ملین پارٹیشنز بنانے کی کوشش نہ کریں۔
- چھانٹنا اب بھی مشکل ہے، جیسا کہ اسپارک کو ترتیب دینا ہے۔
- بعض اوقات خصوصی ڈیٹا کے لیے خصوصی حل کی ضرورت ہوتی ہے۔
- چنگاری جمع کرنا تیز ہے، لیکن تقسیم ابھی بھی مہنگا ہے۔
- جب وہ آپ کو بنیادی باتیں سکھائیں تو نہ سوئیں، شاید کسی نے آپ کا مسئلہ 1980 کی دہائی میں حل کر دیا ہو۔
gnu parallel- یہ ایک جادوئی چیز ہے، ہر ایک کو اسے استعمال کرنا چاہیے؛- چنگاری غیر کمپریسڈ ڈیٹا کو پسند کرتی ہے اور پارٹیشنز کو یکجا کرنا پسند نہیں کرتی ہے۔
- سادہ مسائل کو حل کرتے وقت چنگاری کا سر بہت زیادہ ہوتا ہے۔
- AWK کی ایسوسی ایٹیو صفیں بہت موثر ہیں۔
- آپ رابطہ کر سکتے ہیں
stdinиstdoutR اسکرپٹ سے، اور اس لیے اسے پائپ لائن میں استعمال کریں۔ - سمارٹ پاتھ کے نفاذ کی بدولت، S3 بہت سی فائلوں پر کارروائی کر سکتا ہے۔
- وقت ضائع کرنے کی بنیادی وجہ وقت سے پہلے آپ کے ذخیرہ کرنے کے طریقہ کار کو بہتر بنانا ہے۔
- کاموں کو دستی طور پر بہتر بنانے کی کوشش نہ کریں، کمپیوٹر کو کرنے دیں۔
- استعمال میں آسانی اور لچک کی خاطر API کو آسان ہونا چاہیے۔
- اگر آپ کا ڈیٹا اچھی طرح سے تیار ہے، تو کیشنگ آسان ہو جائے گی!
ماخذ: www.habr.com
