


В текущей пандемии COVID-19 появилось много проблем, на которые хакеры с удовольствием набрасывались. От лицевых щитков, распечатанных на 3D-принтере и медицинских масок домашнего изготовления до замены полноценного механического аппарата искусственной вентиляции лёгких – этот поток идей вдохновлял и радовал душу. В то же самое время были попытки продвинуться и в другой области: в исследованиях, нацеленных на борьбу непосредственно с самим вирусом.
Судя по всему, наибольший потенциал для остановки текущей пандемии и опережения всех последующих есть у подхода, пытающегося докопаться до самого истока проблемы. Этот подход из разряда «узнай своего врага» исповедует вычислительный проект Folding@Home. Миллионы людей зарегистрировались в проекте и жертвуют часть вычислительных мощностей своих процессоров и GPU, создав таким образом крупнейший [распределённый] суперкомпьютер в истории.
Но для чего конкретно используются все эти экзафлопы? Почему нужно бросать такие вычислительные мощности на ? Какая тут работает биохимия, зачем вообще белкам нужно укладываться? Вот краткий обзор фолдинга белков: что это, как он происходит и в чём его важность.
Для начала самое важное: зачем нужны белки?
Белки — жизненно необходимые структуры. Они не только дают строительный материал для клеток, но и служат ферментами-катализаторами практически всех биохимических реакций. Белки, будь они или , представляют собой длинные цепочки , расположенных в определённой последовательности. Функции белков определяются тем, какие аминокислоты расположены в определённых местах белка. Если, к примеру, белку необходимо связываться с положительно заряженной молекулой, место соединения должно быть заполнено отрицательно заряженными аминокислотами.
Чтобы понять, как белки получают структуру, определяющую их функцию, нужно пробежаться по основам молекулярной биологии и информационному потоку в клетке.
Производство, или белков начинается с процесса . Во время транскрипции двойная спираль ДНК, содержащая в себе генетическую информацию клетки, частично расплетается, давая доступ азотных оснований ДНК ферменту под названием . Задача РНК-полимеразы состоит в том, чтобы сделать РНК-копию, или транскрипцию, гена. Эта копия гена под названием (мРНК), представляет собой одинарную молекулу, идеально подходящую для управления внутриклеточными белковыми фабриками, , которые занимаются производством, или белков.
Рибосомы ведут себя как сборочные приспособления – они захватывают шаблон мРНК и сопоставляют его другим небольшим кусочкам РНК, (тРНК). У каждой тРНК есть две активные области – секция из трёх оснований под названием , которая должна совпадать с соответствующими кодонами мРНК, и участок для связывания аминокислоты, специфичной для этого . Во время трансляции молекулы тРНК в рибосоме случайным образом пытаются связаться с мРНК при помощи антикодонов. В случае успеха молекула тРНК присоединяет свою аминокислоту к предыдущей, формируя очередное звено в цепочке аминокислот, закодированной мРНК.
Эта последовательность аминокислот является первым уровнем структурной иерархии белка, поэтому и называется его . Вся трёхмерная структура белка и его функции напрямую происходят от первичной структуры, и зависят от различных свойств каждой из аминокислот и их взаимодействия между собой. Не будь этих химических свойств и взаимодействий аминокислот, так и оставались бы линейными последовательностями без трёхмерной структуры. Это можно увидеть каждый раз во время готовки еды – в этом процессе происходит тепловая трёхмерной структуры белков.
Дальнодействующие связи частей белков
Следующему уровню трёхмерной структуры, выходящему за рамки первичной, дали хитроумное название . В неё входят водородные связи между аминокислотами относительно близкого действия. Основная суть этих стабилизирующих взаимодействий сводится к двум вещам: и . Альфа-спираль образует туго скрученный участок полипептида, а бета-лист – гладкую и широкую область. У обоих образований есть как структурные, так и функциональные свойства, зависящие от характеристик составляющих их аминокислот. К примеру, если альфа-спираль в основном состоит из гидрофильных аминокислот, как или , то она, скорее всего, будет участвовать в водных реакциях.
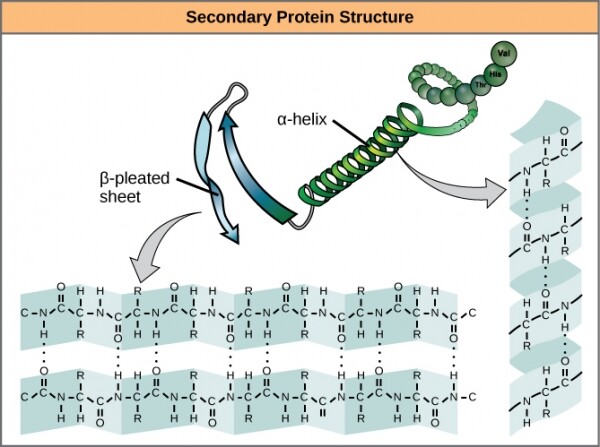
Альфа-спирали и бета-листы в белках. Водородные связи формируются во время экспрессии белка.
Эти две структуры и их комбинации формируют следующий уровень структуры белка — . В отличие от простых фрагментов вторичной структуры, на третичную структуру в основном влияет гидрофобность. В центрах большинства белков содержатся аминокислоты с высокой гидрофобностью, типа или , и вода исключается оттуда из-за «жирной» природы радикалов. Эти структуры часто появляются в трансмембранных белках, встроенных в двойную липидную мембрану, окружающую клетки. Гидрофобные участки белков остаются термодинамически стабильными внутри жировой части мембраны, а гидрофильные участки белка подвергаются воздействию водной среды с обеих её сторон.
Также стабильность третичных структур обеспечивают дальнодействующие связи между аминокислотами. Классическим примером таких связей служит , часто возникающий между двумя радикалами цистеинов. Если в парикмахерской во время процедуры перманентной завивки волос какого-нибудь клиента вы чувствовали запах, немного напоминающей тухлые яйца, то это была частичная денатурация третичной структуры содержащегося в волосах кератина, проходящая посредством уменьшения дисульфидных связей при помощи содержащих серу смесей.
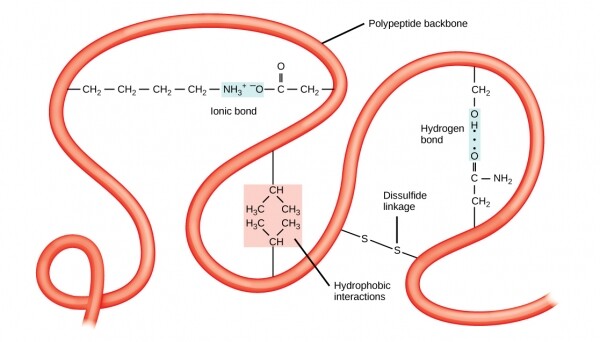
Третичную структуру стабилизируют дальнодействующие взаимодействия, типа гидрофобности или дисульфидных связей
Дисульфидные связи могут возникать между радикалами в одной полипептидной цепочке, или между цистеинами из разных полных цепочек. Взаимодействия между разными цепочками формируют уровень белковой структуры. Прекрасным примером четвертичной структуры служит у вас в крови. Каждая молекула гемоглобина состоит из четырёх одинаковых глобинов, частей белка, каждый из которых удерживается в определённом положении внутри полипептида дисульфидными мостиками, а также связан с молекулой гема, содержащей железо. Все четыре глобина связаны межмолекулярными дисульфидными мостиками, а вся молекула целиком связывается сразу с несколькими молекулами воздуха, вплоть до четырёх, и способна отпускать их по необходимости.
Моделирование структур в поисках лечения болезни
Полипептидные цепочки начинают укладываться в итоговую форму во время трансляции, когда растущая цепочка выходит из рибосомы – примерно как отрезок проволоки из сплава с эффектом памяти может принимать сложные формы при нагреве. Однако, как всегда в биологии, всё не так просто.
Во многих клетках перед трансляцией транскрибированные гены подвергаются серьёзному редактированию, значительно меняющему основную структуру белка по сравнению с чистой последовательностью оснований гена. При этом трансляционные механизмы часто заручаются помощью молекулярных сопровождающих, белков, временно связывающихся с нарождающейся полипептидной цепочкой, и не дающих ей принимать какую-либо промежуточную форму, из которой они потом не смогут перейти к окончательной.
Это всё к тому, что предсказание окончательной формы белка не является тривиальной задачей. Десятилетиями единственным способом изучения структуры белков были физические методы типа рентгеновской кристаллографии. Только в конце 1960-х биофизические химики начали строить вычислительные модели фолдинга белка, в основном сконцентрировавшись на моделировании вторичной структуры. Этим методам и их потомкам требуются огромные объёмы входных данных в дополнение к первичной структуре – к примеру, таблицы углов связи аминокислот, списки гидрофобности, заряженные состояния и даже сохранение структуры и функционирование на эволюционных временных отрезках – и всё для того, чтобы догадаться, как будет выглядеть окончательный белок.
Сегодняшние вычислительные методы предсказания вторичной структуры, работающие, в частности, в сети Folding@Home, работают примерно с 80% точностью – что довольно неплохо, учитывая сложность задачи. Данные, полученные предсказательными моделями по таким белкам, как белок шипов SARS-CoV-2, будут сопоставлены с данными физического изучения вируса. В итоге можно будет получить точную структуру белка и, возможно, разобраться в том, как вирус прикрепляется к рецепторам человека, находящимся в дыхательных путях, ведущих внутрь тела. Если мы сможем разобраться в этой структуре, мы, вероятно, сумеем найти лекарства, блокирующие связывание и предотвращающие инфицирование.
Исследования фолдинга белка лежат в самом сердце нашего понимания такого количества заболеваний и инфекций, что даже когда мы при помощи сети Folding@Home придумаем, как победить COVID-19, за взрывным ростом которого мы наблюдаем в последнее время, эта сеть не будет долго простаивать без работы. Это исследовательский инструмент, отлично подходящий для изучения белковых моделей, лежащих в основе десятков заболеваний, связанных с неправильным фолдингом белков – например, с болезнью Альцгеймера или с разновидностью болезни Крейтцфельдта — Якоба, которую часто некорректно именуют коровьим бешенством. И когда неизбежно появится очередной вирус, мы уже будем готовы снова начать с ним борьбу.
Источник: habr.com
