


Как читать эту статью: прошу прощения за то, что текст получился таким длинным и хаотичным. Чтобы сэкономить ваше время, я каждую главу начинаю со вступления «Чему я научился», в котором одним-двумя предложениями излагаю суть главы.
«Просто покажи решение!» Если вы хотите всего лишь увидеть, к чему я пришёл, то переходите к главе «Становлюсь изобретательнее», но я считаю, что интереснее и полезнее почитать про неудачи.
Недавно мне поручили настроить процесс обработки большого объёма исходных последовательностей ДНК (технически это SNP-чип). Нужно было быстро получать данные о заданном генетическом местоположении (которое называется SNP) для последующего моделирования и прочих задач. С помощью R и AWK мне удалось очистить и организовать данные естественным образом, сильно ускорив обработку запросов. Далось мне это нелегко и потребовало многочисленных итераций. Эта статья поможет вам избежать некоторых моих ошибок и продемонстрирует, что же у меня в конце концов получилось.
Для начала некоторые вводные пояснения.
Данные
Наш университетский центр обработки генетической информации предоставил нам данные в виде TSV объёмом 25 Тб. Мне они достались разбитыми на 5 пакетов, сжатых Gzip, каждый из которых содержал около 240 четырёхгигабайтных файлов. Каждый ряд содержал данные для одного SNP одного человека. Всего были переданы данные по ~2,5 млн SNP и ~60 тыс. человек. Кроме SNP-информации в файлах были многочисленные колонки с числами, отражающими различные характеристики, такие как интенсивность чтения, частота разных аллелей и т.д. Всего было порядка 30 колонок с уникальными значениями.
Цель
Как и в любом проекте по управлению данными, самым важным было определить, как будут использоваться данные. В этом случае мы по большей части будем подбирать модели и рабочие процессы для SNP на основе SNP. То есть одновременно нам будут нужны данные только по одному SNP. Я должен был научиться как можно проще, быстрее и дешевле извлекать все записи, относящиеся к одному из 2,5 миллионов SNP.
Как этого не делать
Процитирую подходящее клише:
Я не терпел тысячу раз неудачу, я лишь открыл тысячу способов не парсить кучу данных в формате, удобном для запросов.
Первая попытка
Чему я научился: не существует дешёвого способа отпарсить 25 Тб за раз.
Прослушав в Университете Вандербильта предмет «Расширенные методы обработки больших данных», я был уверен, что дело в шляпе. Пожалуй, час-два уйдёт на настройку Hive-сервера, чтобы пробежать по всем данным и отчитаться о результате. Поскольку наши данные хранятся в AWS S3, я воспользовался сервисом , который позволяет применять Hive SQL-запросы к S3-данным. Не надо настраивать/поднимать Hive-кластер, да ещё и платишь только за те данные, которые ищешь.
После того как я показал Athena свои данные и их формат, я прогнал несколько тестов с подобными запросами:
select * from intensityData limit 10;И быстро получил хорошо структурированные результаты. Готово.
Пока мы не попробовали использовать данные в работе…
Меня попросили вытащить всю информацию по SNP, чтобы протестировать на ней модель. Я запустил запрос:
select * from intensityData
where snp = 'rs123456';…и стал ждать. Спустя восемь минут и больше 4 Тб запрошенных данных я получил результат. Athena берёт плату за объём найденных данных, по $5 за терабайт. Так что этот единственный запрос обошёлся в $20 и восемь минут ожидания. Чтобы прогнать модель по всем данным, нужно было прождать 38 лет и заплатить $50 млн. Очевидно, что нам это не подходило.
Нужно было использовать Parquet…
Чему я научился: будьте осторожны с размером ваших Parquet-файлов и их организацией.
Сначала я попытался исправить ситуацию, конвертировав все TSV в . Они удобны для работы с большими наборами данных, потому что информация в них хранится в колоночном виде: каждая колонка лежит в собственном сегменте памяти/диска, в отличие от текстовых файлов, в которых строки содержат элементы каждой колонки. И если нужно что-то найти, то достаточно прочитать необходимую колонку. Кроме того, в каждом файле в колонке хранится диапазон значений, так что если искомое значение отсутствует в диапазоне колонки, Spark не будет тратить время на сканирование всего файла.
Я запустил простую задачу для преобразования наших TSV в Parquet и забросил новые файлы в Athena. Это заняло около 5 часов. Но когда я запустил запрос, то на его выполнение ушло примерно столько же времени и чуть меньше денег. Дело в том, что Spark, пытаясь оптимизировать задачу, просто распаковал один TSV-чанк и положил его в собственный Parquet-чанк. И поскольку каждый чанк был достаточно велик и вмещал полные записи многих людей, то в каждом файле хранились все SNP, поэтому Spark’у приходилось открывать все файлы, чтобы извлечь нужную информацию.
Любопытно, что используемый по умолчанию (и рекомендуемый) тип компрессии в Parquet — snappy, — не является разделяемым (splitable). Поэтому каждый исполнитель (executor) залипал на задаче распаковки и загрузи полного датасета на 3,5 Гб.
Разбираемся в проблеме
Чему я научился: сортировать сложно, особенно, если данные распределены.
Мне казалось, что теперь я понял суть проблемы. Мне нужно было лишь отсортировать данные по колонке SNP, а не по людям. Тогда в отдельном чанке данных будет храниться несколько SNP, и тогда себя во всей красе проявит «умная» функция Parquet «открывать только если значение находится в диапазоне». К сожалению, отсортировать миллиарды строк, раскиданных по кластеру, оказалось сложной задачей.
Me taking algorithms class in college: «Ugh, no one cares about computational complexity of all these sorting algorithms»
Me trying to sort on a column in a 20TB table: «Why is this taking so long?» struggles.
— Nick Strayer (@NicholasStrayer)
AWS точно не хочет возвращать деньги по причине «Я рассеянный студент». После того, как я запустил сортировку на Amazon Glue, она проработала 2 дня и завершилась сбоем.
Что насчёт партиционирования?
Чему я научился: партиции в Spark должны быть сбалансированы.
Затем мне пришла в голову идея партиционировать данные в хромосомах. Их 23 штуки (и ещё несколько, если учитывать митохондриальную ДНК и нерасшифрованные (unmapped) области).
Это позволит разделить данные на более мелкие порции. Если добавить в Spark-функцию экспорта в скрипте Glue всего лишь одну строку partition_by = "chr", то данные должны быть разложены по бакетам (buckets).
Геном состоит из многочисленных фрагментов, которые называются хромосомами.
К сожалению, это не сработало. У хромосом разные размеры, а значит и разное количество информации. Это значит, задачи, которые Spark отправлял воркерам, не были сбалансированы и выполнялись медленно, потому что некоторые узлы заканчивали раньше и простаивали. Однако задачи были выполнены. Но при запросе одного SNP несбалансированность снова стала причиной проблем. Стоимость обработки SNP в более крупных хромосомах (то есть там, откуда мы и хотим получить данные) уменьшилась всего лишь примерно в 10 раз. Много, но не достаточно.
А если разделить на ещё более мелкие партиции?
Чему я научился: вообще никогда не пытайтесь делать 2,5 миллиона партиций.
Я решил гулять по полной и партиционировал каждый SNP. Это гарантировало одинаковый размер партиций. ПЛОХАЯ БЫЛА ИДЕЯ. Я воспользовался Glue и добавил невинную строку partition_by = 'snp'. Задача запустилась и начала выполняться. День спустя я проверил, и увидел, что в S3 до сих пор ничего не записано, поэтому убил задачу. Похоже, Glue записывал промежуточные файлы в скрытое место в S3, и много файлов, возможно, пару миллионов. В результате моя ошибка обошлась более чем в тысячу долларов и не обрадовала моего наставника.
Партиционирование + сортировка
Чему я научился: сортировать всё ещё трудно, как и настраивать Spark.
Последняя попытка партиционирования заключалась в том, что я партиционировал хромосомы, а затем сортировал каждую партицию. В теории, это позволяло бы ускорить каждый запрос, потому что желаемые данные об SNP должны были находиться в пределах нескольких Parquet-чанков внутри заданного диапазона. Увы, сортировка даже партиционированных данных оказалась трудной задачей. В результате я перешёл на EMR для кастомного кластера и использовал восемь мощных инстансов (C5.4xl) и Sparklyr для создания более гибкого рабочего процесса…
# Sparklyr snippet to partition by chr and sort w/in partition
# Join the raw data with the snp bins
raw_data
group_by(chr) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr')
)…однако задача всё равно так и не была выполнена. Я настраивал по-всякому: увеличивал выделение памяти для каждого исполнителя запросов, использовал узлы с большим объёмом памяти, применял широковещательные переменные (broadcasting variable), но каждый раз это оказывались полумеры, и постепенно исполнители начинали сбоить, пока всё не остановилось.
Update: so it begins.
— Nick Strayer (@NicholasStrayer)
Становлюсь изобретательнее
Чему я научился: иногда особые данные требуют особых решений.
У каждого SNP есть значение позиции. Это число, соответствующее количеству оснований, лежащих вдоль его хромосомы. Это хороший и естественный способ организации наших данных. Сначала я хотел партиционировать по областям каждой хромосомы. Например, позиции 1 — 2000, 2001 — 4000 и т.д. Но проблема в том, что SNP распределены по хромосомам неравномерно, поэтому поэтому размер групп будет сильно различаться.
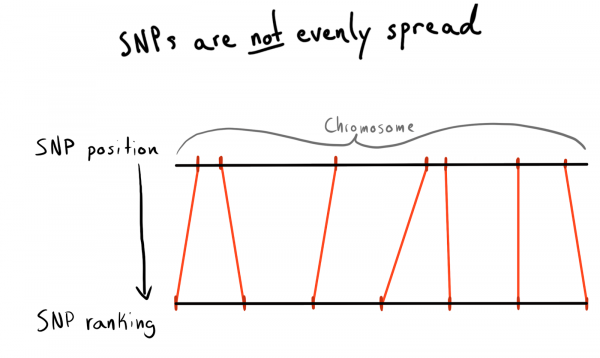
В результате я пришёл к разбиению по категориям (rank) позиций. По уже загруженным данным я прогнал запрос на получение списка уникальных SNP, их позиций и хромосом. Затем отсортировал данные внутри каждой хромосомы и собрал SNP в группы (bin) заданного размера. Скажем, по 1000 SNP. Это дало мне взаимосвязь SNP с группой-в-хромосоме.
В конце концов я сделал группы (bin) по 75 SNP, причину объясню ниже.
snp_to_bin <- unique_snps %>%
group_by(chr) %>%
arrange(position) %>%
mutate(
rank = 1:n()
bin = floor(rank/snps_per_bin)
) %>%
ungroup()Первая попытка со Spark
Чему я научился: объединение в Spark работает быстро, но партиционирование всё ещё обходится дорого.
Я хотел прочитать этот маленький (2,5 млн строк) фрейм данных в Spark, объединить его с сырыми данными, а затем партиционировать по свежедобавленной колонке bin.
# Join the raw data with the snp bins
data_w_bin <- raw_data %>%
left_join(sdf_broadcast(snp_to_bin), by ='snp_name') %>%
group_by(chr_bin) %>%
arrange(Position) %>%
Spark_write_Parquet(
path = DUMP_LOC,
mode = 'overwrite',
partition_by = c('chr_bin')
) Я использовал sdf_broadcast(), таким образом Spark узнаёт, что он должен отправить фрейм данных во все узлы. Это полезно, если данные небольшого размера и требуются для всех задач. Иначе Spark пытается быть умным и распределяет данные по мере надобности, что может стать причиной тормозов.
И опять моя задумка не сработала: задачи какое-то время работали, завершали объединение, а затем, как и запущенные партиционированием исполнители, начали сбоить.
Добавляю AWK
Чему я научился: не спите, когда вам преподают основы. Наверняка кто-то уже решил вашу проблему ещё в 1980-х.
До этого момента причиной всех моих неудач со Spark была перемешанность данных в кластере. Возможно, ситуацию можно улучшить с помощью предварительной обработки. Я решил попробовать разделить сырые текстовые данные на колонки хромосом, так я надеялся предоставить Spark’у «предварительно партиционированные» данные.
Я поискал на StackOverflow, как разбивать по значениям колонок, и нашёл С помощью AWK вы можете разделить текстовый файл по значениям колонок, выполнив запись в скрипте, а не отправляя результаты в stdout.
Для пробы я написал Bash-скрипт. Скачал один из запакованных TSV, затем распаковал его с помощью gzip и отправил в awk.
gzip -dc path/to/chunk/file.gz |
awk -F 't'
'{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'Это сработало!
Заполнение ядер
Чему я научился: gnu parallel — это волшебная вещь, все должны её использовать.
Разделение проходило довольно медленно, и когда я запустил htop, чтобы проверить использование мощного (и дорогого) EC2-инстанса, то оказалось, что я задействую только одно ядро и примерно 200 Мб памяти. Чтобы решить задачу и не потерять кучу денег, нужно было придумать, как распараллелить работу. К счастью, в совершенно потрясающей книге Джерона Джанссенса я нашёл главу, посвящённую распараллеливанию. Из неё я узнал про gnu parallel, очень гибкий метод реализации многопоточности в Unix.

Когда я запустил разделение с помощью нового процесса, всё было прекрасно, но оставалось узкое место — скачивание S3-объектов на диск было не слишком быстрым и не полностью распараллеленным. Чтобы это исправить, я сделал вот что:
- Выяснил, что можно прямо в конвейере реализовать этап S3-скачивания, полностью исключив промежуточное хранение на диске. Это означает, что я могу избежать записи сырых данных на диск и использовать ещё более маленькое, а значит и более дешёвое хранилище на AWS.
- Командой
aws configure set default.s3.max_concurrent_requests 50сильно увеличил количество потоков, которые использует AWS CLI (по умолчанию их 10). - Перешёл на оптимизированный по скорости сети инстанс EC2, с буквой n в названии. Я обнаружил, что потеря вычислительной мощности при использовании n-инстансов с лихвой компенсируется увеличением скорости загрузки. Для большинства задач я использовал c5n.4xl.
- Поменял
gzipна , это gzip-инструмент, который умеет делать классные штуки для распараллеливания изначально не распараллеленной задачи распаковки файлов (это помогло меньше всего).
# Let S3 use as many threads as it wants
aws configure set default.s3.max_concurrent_requests 50
for chunk_file in $(aws s3 ls $DATA_LOC | awk '{print $4}' | grep 'chr'$DESIRED_CHR'.csv') ; do
aws s3 cp s3://$batch_loc$chunk_file - |
pigz -dc |
parallel --block 100M --pipe
"awk -F 't' '{print $1",..."$30">"chunked/{#}_chr"$15".csv"}'"
# Combine all the parallel process chunks to single files
ls chunked/ |
cut -d '_' -f 2 |
sort -u |
parallel 'cat chunked/*_{} | sort -k5 -n -S 80% -t, | aws s3 cp - '$s3_dest'/batch_'$batch_num'_{}'
# Clean up intermediate data
rm chunked/*
doneЭти шаги скомбинированы друг с другом, чтобы всё работало очень быстро. Благодаря увеличению скорости скачивания и отказу от записи на диск я теперь мог обработать 5-терабайтный пакет всего за несколько часов.
There’s nothing sweeter than seeing all the cores you’re paying for on AWS being used. Thanks to gnu-parallel I can unzip and split a 19gig csv just as fast as I can download it. I couldn’t even get spark to run this.
— Nick Strayer (@NicholasStrayer)
В этом твите должно было упоминаться ‘TSV’. Увы.
Использование заново отпарсенных данных
Чему я научился: Spark любит несжатые данные и не любит комбинировать партиции.
Теперь данные лежали в S3 в незапакованном (читай, разделяемом) и полуупорядоченном формате, и я мог снова вернуться к Spark’у. Меня ждал сюрприз: мне опять не удалось добиться желаемого! Было очень сложно точно сказать Spark’у, как партиционированы данные. И даже когда я это сделал, оказалось, что партиций слишком много (95 тыс.), и когда я с помощью coalesce уменьшил их количество до разумных пределов, это порушило моё партиционирование. Уверен, это можно исправить, но за пару дней поисков мне не удалось найти решение. В конце концов я доделал все задачи в Spark, хотя это заняло какое-то время, а мои разделённые Parquet-файлы были не очень маленькими (~200 Кб). Однако данные лежали там, где нужно.
Слишком маленькие и неодинаковые, чудесно!
Тестирование локальных Spark-запросов
Чему я научился: в Spark слишком много накладных расходов при решении простых задач.
Загрузив данные в продуманном формате, я смог протестировать скорость. Настроил скрипт на R для запуска локального Spark-сервера, а потом загрузил фрейм данных Spark из указанного хранилища Parquet-групп (bin). Я пытался загрузить все данные, но не смог заставить Sparklyr распознать партиционирование.
sc <- Spark_connect(master = "local")
desired_snp <- 'rs34771739'
# Start a timer
start_time <- Sys.time()
# Load the desired bin into Spark
intensity_data <- sc %>%
Spark_read_Parquet(
name = 'intensity_data',
path = get_snp_location(desired_snp),
memory = FALSE )
# Subset bin to snp and then collect to local
test_subset <- intensity_data %>%
filter(SNP_Name == desired_snp) %>%
collect()
print(Sys.time() - start_time)Исполнение заняло 29,415 секунды. Гораздо лучше, но не слишком хорошо для массового тестирования чего-либо. Кроме того, я не мог ускорить работу с помощью кэширования, потому что когда я пытался кэшировать в памяти фрейм данных, Spark всегда падал, даже когда я выделил больше 50 Гб памяти для датасета, который весил меньше 15.
Возвращение к AWK
Чему я научился: ассоциативные массивы в AWK очень эффективны.
Я понимал, что могу добиться более высокой скорости. Мне вспомнилось, что в замечательном я читал о клёвой фиче, которая называется «». По сути, это пары ключ-значение, которые почему-то в AWK назвали иначе, и поэтому я как-то о них и не вспоминал особо. напомнил, что термин «ассоциативные массивы» гораздо старше термина «пара ключ-значение». Даже если вы , этот термин вы там не увидите, зато найдёте ассоциативные массивы! К тому же «пара ключ-значение» чаще всего ассоциируется базами данных, поэтому гораздо логичнее сравнивать с hashmap. Я понял, что могу использовать эти ассоциативные массивы для связи моих SNP с таблицей групп (bin table) и сырыми данными без применения Spark.
Для этого в AWK-скрипте я использовал блок BEGIN. Это фрагмент кода, который исполняется до того, как первая строка данных будет передана в основное тело скрипта.
join_data.awk
BEGIN {
FS=",";
batch_num=substr(chunk,7,1);
chunk_id=substr(chunk,15,2);
while(getline < "snp_to_bin.csv") {bin[$1] = $2}
}
{
print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv"
} Команда while(getline...) загрузила все строки из CSV группы (bin), задала первую колонку (название SNP) в качестве ключа для ассоциативного массива bin и второе значение (группа) в качестве значения. Затем в блоке { }, который исполняется применительно ко всем строкам основного файла, каждая строка отправляется в выходной файл, получающий уникальное имя в зависимости от его группы (bin): ..._bin_"bin[$1]"_....
Переменные batch_num и chunk_id соответствовали данным, предоставленным конвейером, что позволило избежать состояния гонки, и каждый поток исполнения, запущенный parallel, писал в собственный уникальный файл.
Поскольку все сырые данные я раскидал по папкам по хромосомам, оставшимся после моего предыдущего эксперимента с AWK, теперь я мог написать другой Bash-скрипт, чтобы обрабатывать по хромосоме за раз и отдавать в S3 глубже партиционированные данные.
DESIRED_CHR='13'
# Download chromosome data from s3 and split into bins
aws s3 ls $DATA_LOC |
awk '{print $4}' |
grep 'chr'$DESIRED_CHR'.csv' |
parallel "echo 'reading {}'; aws s3 cp "$DATA_LOC"{} - | awk -v chr=""$DESIRED_CHR"" -v chunk="{}" -f split_on_chr_bin.awk"
# Combine all the parallel process chunks to single files and upload to rds using R
ls chunked/ |
cut -d '_' -f 4 |
sort -u |
parallel "echo 'zipping bin {}'; cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R '$S3_DEST'/chr_'$DESIRED_CHR'_bin_{}.rds"
rm chunked/* В скрипте два раздела parallel.
В первом разделе считываются данные из всех файлов, содержащих информацию по нужной хромосоме, затем эти данные распределяются по потокам, которые раскидывают файлы по соответствующим группам (bin). Чтобы не возникало состояния гонки, когда несколько потоков записывают в один файл, AWK передаёт имена файлов для записи данных в разные места, например, chr_10_bin_52_batch_2_aa.csv. В результате на диске создаётся множество маленьких файлов (для этого я использовал терабайтные EBS-тома).
Конвейер из второго раздела parallel проходит по группам (bin) и объединяет их отдельные файлы в общие CSV c cat, а затем отправляет их на экспорт.
Транслирование в R?
Чему я научился: можно обращаться к stdin и stdout из R-скрипта, а значит и использовать его в конвейере.
В Bash-скрипте вы могли заметить такую строку: ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R.... Она транслирует все конкатенированные файлы группы (bin) в нижеприведённый R-скрипт. {} является специальной методикой parallel, которая любые отправляемые ею в указанный поток данные вставляет в прямо в саму команду. Опция {#} предоставляет уникальный ID потока исполнения, а {%} представляет собой номер слота задания (повторяется, но никогда одновременно). Список всех опций можно найти в
#!/usr/bin/env Rscript
library(readr)
library(aws.s3)
# Read first command line argument
data_destination <- commandArgs(trailingOnly = TRUE)[1]
data_cols <- list(SNP_Name = 'c', ...)
s3saveRDS(
read_csv(
file("stdin"),
col_names = names(data_cols),
col_types = data_cols
),
object = data_destination
) Когда переменная file("stdin") передаётся в readr::read_csv, данные, транслированные в R-скрипт, загружаются во фрейм, который потом в виде .rds-файла с помощью aws.s3 записывается прямо в S3.
RDS — это что-то вроде младшей версии Parquet, без изысков колоночного хранилища.
После завершения Bash-скрипта я получил пачку .rds-файлов, лежащих в S3, что дало позволило мне использовать эффективное сжатие и встроенные типы.
Несмотря на использование тормозного R, всё работало очень быстро. Неудивительно, что фрагменты на R, отвечающие за чтение и запись данных, хорошо оптимизированы. После тестирования на одной хромосоме среднего размера, задание выполнилось на инстансе C5n.4xl примерно за два часа.
Ограничения S3
Чему я научился: благодаря умной реализации путей S3 может обрабатывать много файлов.
Я беспокоился, сможет ли S3 обработать множество переданных ей файлов. Я мог сделать имена файлов осмысленными, но как S3 будет по ним искать?

Папки в S3 всего лишь для красоты, на самом деле систему не интересует символ /.
Похоже, S3 представляет путь до конкретного файла в виде простого ключа в своеобразной хеш-таблице или базе данных на основе документов. Бакет (bucket) можно считать таблицей, а файлы — записями в этой таблице.
Поскольку скорость и эффективность важны для получения прибыли в Amazon, неудивительно, что эта система «ключ-в-качестве-пути-к-файлу» офигенно оптимизирована. Я пытался найти баланс: чтобы не нужно было делать множество get-запросов, но чтобы при этом запросы выполнялись быстро. Оказалось, что лучше всего делать около 20 тыс. bin-файлов. Думаю, если продолжить оптимизировать, то можно добиться повышения скорости (например, делать специальный бакет только для данных, таким образом уменьшая размер поисковой таблицы). Но на дальнейшие эксперименты уже не было времени и денег.
Что насчёт перекрёстной совместимости?
Чему я научился: главная причина потери времени — преждевременная оптимизация вашего метода хранения.
В этот момент очень важно спросить себя: «Зачем использовать проприетарный формат файлов?» Причина кроется в скорости загрузки (запакованные gzip CSV-файлы грузились в 7 раз дольше) и совместимости с нашими рабочими процессами. Я могу пересмотреть своё решение, если R сможет легко загружать файлы Parquet (или Arrow) без нагрузки в виде Spark. У нас в лаборатории все используют R, и если мне понадобится преобразовать данные в другой формат, то у меня всё ещё остаются исходные текстовые данные, поэтому я могу просто снова запустить конвейер.
Разделение работы
Чему я научился: не пытайтесь оптимизировать задания вручную, пусть это делает компьютер.
Я отладил рабочий процесс на одной хромосоме, теперь нужно обработать все остальные данные.
Хотелось поднять несколько инстансов EC2 для преобразования, но в то же время я опасался получить крайне несбалансированную нагрузку в разных заданиях на обработку (так же, как Spark страдал от несбалансированных партиций). Кроме того, мне не улыбалось поднимать по одному инстансу на каждую хромосому, потому что для AWS-аккаунтов есть ограничение по умолчанию в 10 инстансов.
Тогда я решил написать на R скрипт для оптимизации заданий на обработку.
Сначала попросил S3 вычислить, сколько места в хранилище занимает каждая хромосома.
library(aws.s3)
library(tidyverse)
chr_sizes <- get_bucket_df(
bucket = '...', prefix = '...', max = Inf
) %>%
mutate(Size = as.numeric(Size)) %>%
filter(Size != 0) %>%
mutate(
# Extract chromosome from the file name
chr = str_extract(Key, 'chr.{1,4}.csv') %>%
str_remove_all('chr|.csv')
) %>%
group_by(chr) %>%
summarise(total_size = sum(Size)/1e+9) # Divide to get value in GB
# A tibble: 27 x 2
chr total_size
<chr> <dbl>
1 0 163.
2 1 967.
3 10 541.
4 11 611.
5 12 542.
6 13 364.
7 14 375.
8 15 372.
9 16 434.
10 17 443.
# … with 17 more rows Затем я написал функцию, которая берёт общий размер, перемешивает порядок хромосом, делит их на группы num_jobs и сообщает, насколько различаются размеры всех заданий на обработку.
num_jobs <- 7
# How big would each job be if perfectly split?
job_size <- sum(chr_sizes$total_size)/7
shuffle_job <- function(i){
chr_sizes %>%
sample_frac() %>%
mutate(
cum_size = cumsum(total_size),
job_num = ceiling(cum_size/job_size)
) %>%
group_by(job_num) %>%
summarise(
job_chrs = paste(chr, collapse = ','),
total_job_size = sum(total_size)
) %>%
mutate(sd = sd(total_job_size)) %>%
nest(-sd)
}
shuffle_job(1)
# A tibble: 1 x 2
sd data
<dbl> <list>
1 153. <tibble [7 × 3]>Потом я прогнал с помощью purrr тысячу перемешиваний и выбрал лучшее.
1:1000 %>%
map_df(shuffle_job) %>%
filter(sd == min(sd)) %>%
pull(data) %>%
pluck(1) Так я получил набор заданий, очень похожих по размеру. Затем оставалось только обернуть мой предыдущий Bash-скрипт в большой цикл for. На написание этой оптимизации ушло около 10 минут. И это куда меньше, чем я потратил бы на ручное создание заданий в случае с их несбалансированностью. Поэтому считаю, что с этой предварительной оптимизацией я не прогадал.
for DESIRED_CHR in "16" "9" "7" "21" "MT"
do
# Code for processing a single chromosome
fiВ конце добавляю команду выключения:
sudo shutdown -h now … и всё получилось! С помощью AWS CLI я поднимал инстансы и через опцию user_data передавал им Bash-скрипты их заданий на обработку. Они исполнялись и автоматически выключались, так что я не платил за излишнюю вычислительную мощность.
aws ec2 run-instances ...
--tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]"
--user-data file://<<job_script_loc>>Упаковываем!
Чему я научился: API должен быть простым ради простоты и гибкости использования.
Наконец-то я получил данные в нужном месте и виде. Оставалось максимально упростить процесс использования данных, чтобы моим коллегам было легче. Я хотел сделать простой API для создания запросов. Если в будущем я решу перейти с .rds на Parquet-файлы, то это должно быть проблемой для меня, а не для коллег. Для этого я решил сделать внутренний R-пакет.
Собрал и задокументировал очень простой пакет, содержащий всего несколько функций для доступа к данным, собранных вокруг функции get_snp. Также я сделал для коллег сайт , чтобы они могли легко посмотреть примеры и документацию.
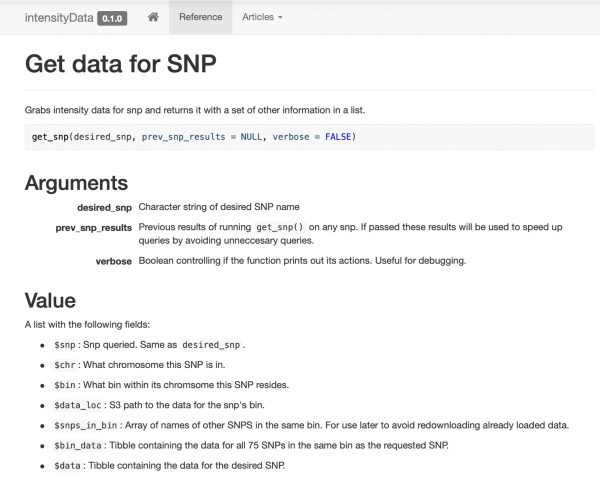
Интеллектуальное кэширование
Чему я научился: если ваши данные хорошо подготовлены, кэшировать будет просто!
Поскольку один из основных рабочих процессов применял к пакету SNP одну и ту же модель анализа, я решил использовать группирование (binning) в своих интересах. При передаче данных по SNP, к возвращаемому объекту прикрепляется и вся информация из группы (bin). То есть старые запросы могут (в теории) ускорять обработку новых запросов.
# Part of get_snp()
...
# Test if our current snp data has the desired snp.
already_have_snp <- desired_snp %in% prev_snp_results$snps_in_bin
if(!already_have_snp){
# Grab info on the bin of the desired snp
snp_results <- get_snp_bin(desired_snp)
# Download the snp's bin data
snp_results$bin_data <- aws.s3::s3readRDS(object = snp_results$data_loc)
} else {
# The previous snp data contained the right bin so just use it
snp_results <- prev_snp_results
}
... При сборке пакета я прогнал много бенчмарков, чтобы сравнить скорость при использовании разных методов. Рекомендую этим не пренебрегать, потому что иногда результаты бывают неожиданными. Например, dplyr::filter оказался гораздо быстрее захвата строк с помощью фильтрации на основе индексирования, а получение одной колонки из отфильтрованного фрейма данных работало гораздо быстрее, чем применение синтаксиса индексирования.
Обратите внимание, что объект prev_snp_results содержит ключ snps_in_bin. Это массив всех уникальных SNP в группе (bin), позволяющий быстро проверять, есть ли уже данные из предыдущего запроса. Также он упрощает циклический проход по всем SNP в группе (bin) с помощью этого кода:
# Get bin-mates
snps_in_bin <- my_snp_results$snps_in_bin
for(current_snp in snps_in_bin){
my_snp_results <- get_snp(current_snp, my_snp_results)
# Do something with results
}Результаты
Теперь мы можем (и начали всерьёз) прогонять модели и сценарии, ранее нам недоступные. Самое лучшее, что моим коллегам по лаборатории не приходится думать о всяких сложностях. У них просто есть работающая функция.
И хотя пакет избавляет их от подробностей, я пытался сделать формат данных достаточно простым, чтобы в нём могли разобраться, если завтра я вдруг исчезну…
Скорость заметно выросла. Обычно мы сканируем функционально значимые фрагменты генома. Раньше мы не могли это делать (получалось слишком дорого), но теперь, благодаря групповой (bin) структуре и кэшированию, на запрос одного SNP уходит в среднем меньше 0,1 секунды, а использование данных такое низкое, что расходы на получаются S3 копеечные.
Recently I got put in change of wrangling 25+ TB of raw genotyping data for my lab. When I started, using spark took 8 min & cost $20 to query a SNP. After using AWK + to process, it now takes less than a 10th of a second and costs $0.00001. My personal win.
— Nick Strayer (@NicholasStrayer)
Заключение
Эта статья — совсем не руководство. Решение получилось индивидуальное, и почти наверняка не оптимальное. Скорее, это рассказ о путешествии. Хочу, чтобы другие поняли, что подобные решения не возникают в голове полностью сформированными, это результат проб и ошибок. Кроме того, если вы ищете специалиста по анализу данных, то учитывайте, что для эффективного использования этих инструментов нужен опыт, а опыт требует денег. Я счастлив, что у меня были средства на оплату, но у многих других, кто может сделать ту же работу лучше меня, никогда не будет такой возможности из-за отсутствия денег даже на попытку.
Инструменты для больших данных универсальны. Если у вас есть время, то почти наверняка сможете написать более быстрое решение, применив «умную» очистку данных, хранилище и методики извлечения. В конечном счете все сводится к анализу расходов и выгод.
Чему я научился:
- не существует дешёвого способа отпарсить 25 Тб за раз;
- будьте осторожны с размером ваших Parquet-файлов и их организацией;
- партиции в Spark должны быть сбалансированы;
- вообще никогда не пытайтесь делать 2,5 миллиона партиций;
- сортировать всё ещё трудно, как и настраивать Spark;
- иногда особые данные требуют особых решений;
- объединение в Spark работает быстро, но партиционирование всё ещё обходится дорого;
- не спите, когда вам преподают основы, наверняка кто-то уже решил вашу проблему ещё в 1980-х;
gnu parallel— это волшебная вещь, все должны её использовать;- Spark любит несжатые данные и не любит комбинировать партиции;
- в Spark слишком много накладных расходов при решении простых задач;
- ассоциативные массивы в AWK очень эффективны;
- можно обращаться к
stdinиstdoutиз R-скрипта, а значит и использовать его в конвейере; - благодаря умной реализации путей S3 может обрабатывать много файлов;
- главная причина потери времени — преждевременная оптимизация вашего метода хранения;
- не пытайтесь оптимизировать задания вручную, пусть это делает компьютер;
- API должен быть простым ради простоты и гибкости использования;
- если ваши данные хорошо подготовлены, кэшировать будет просто!
Источник: habr.com
